Miliotou Androulla N, Papagiannopoulou Dionysia, Vlachaki Efthymia, Samiotaki Martina, Laspa Dimitra, Theodoridou Stamatia, Tsiftsoglou Asterios S, Papadopoulou Lefkothea C
Laboratory of Pharmacology, Department of Pharmacognosy - Pharmacology, School of Pharmacy, Faculty of Health Sciences, Aristotle University of Thessaloniki, 54124, Thessaloniki, Macedonia, Greece.
Department of Pharmaceutical Chemistry, School of Pharmacy, Aristotle University of Thessaloniki, 54124, Thessaloniki, Macedonia, Greece.
J Biol Res (Thessalon). 2021 Jul 20;28(1):16. doi: 10.1186/s40709-021-00148-3.
α-Thalassemia, a congenital hemoglobinopathy, is characterized by deficiency and/or reduced levels of α-globin chains in serious forms of α-thalassemia (HbH disease/Hb Bart's). This research work deals with a Protein Replacement Therapy approach in order to manage α-thalassemia manifestations, caused by the excess of β-globin chain into HbH RBCs. The main goal was to produce the recombinant human α-globin chain in fusion with TAT, a Protein Transduction Domain, to ex vivo deliver it into HbH patients RBCs, to replace the endogenous missing α-globin chain.
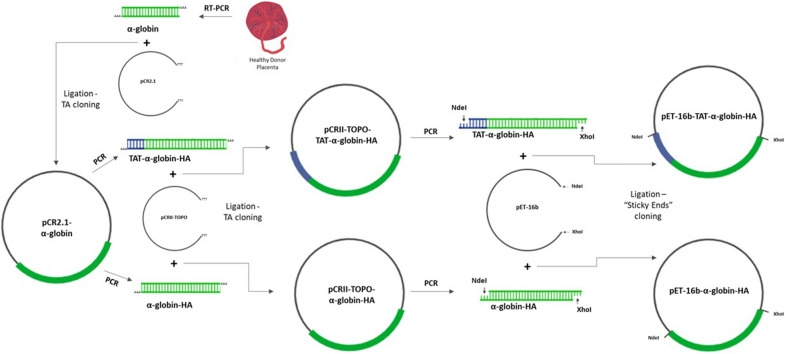
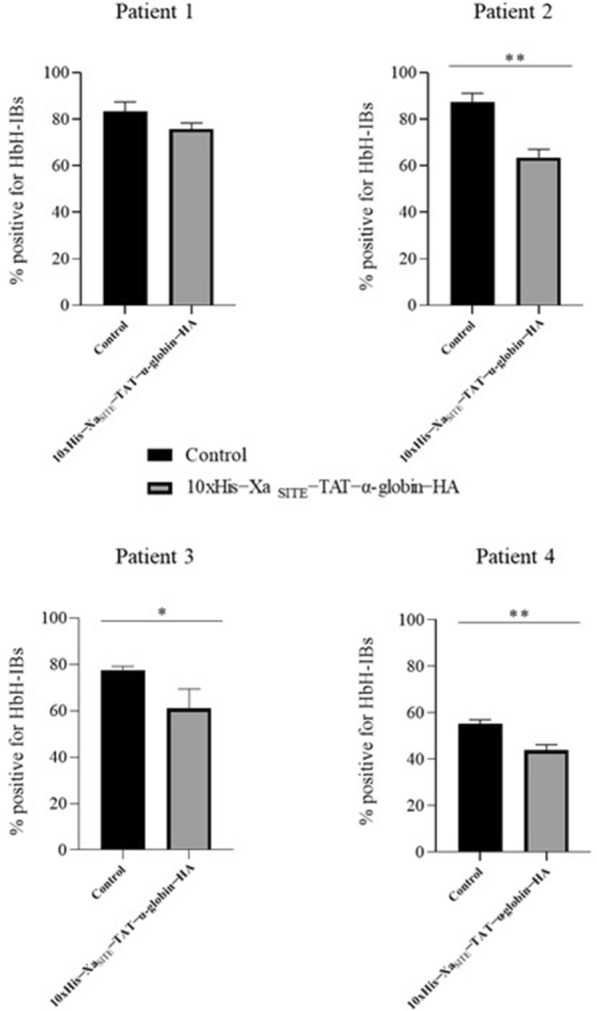
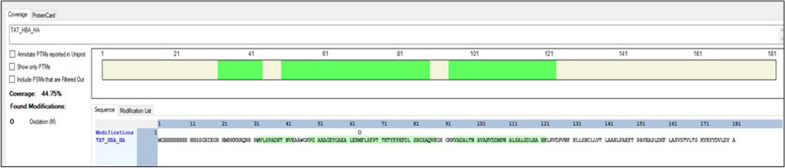
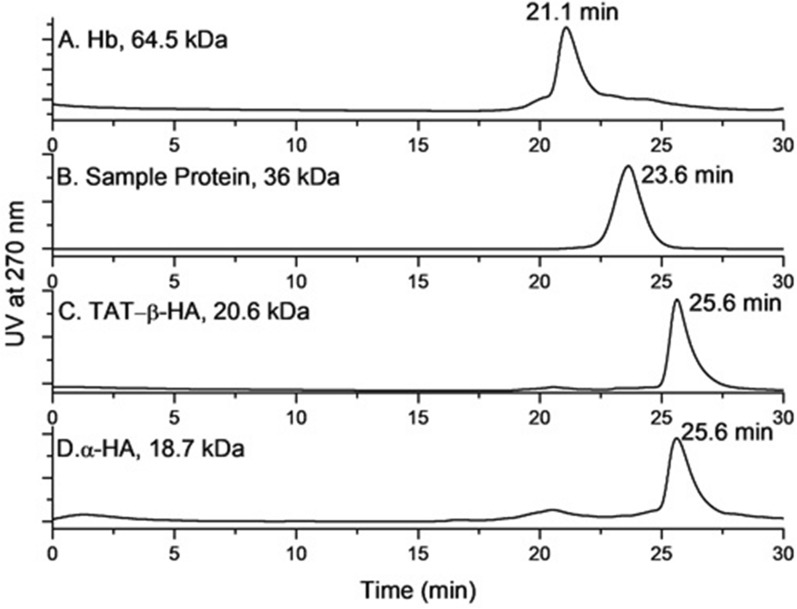
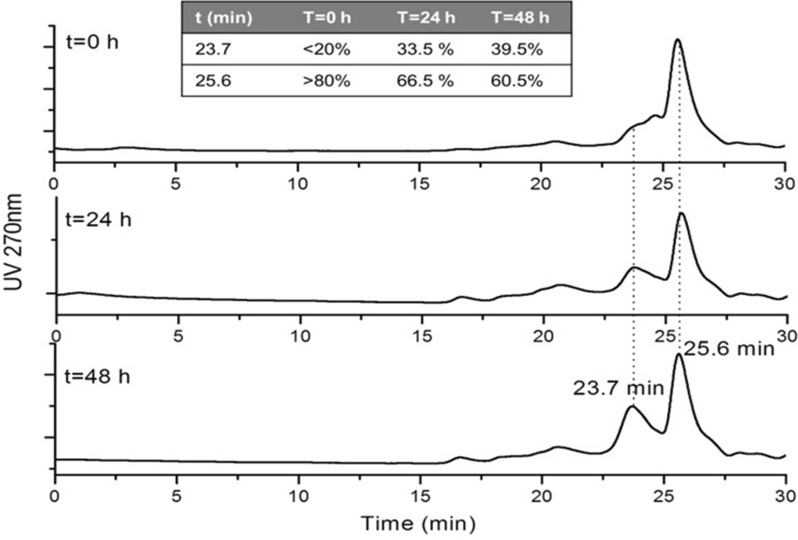
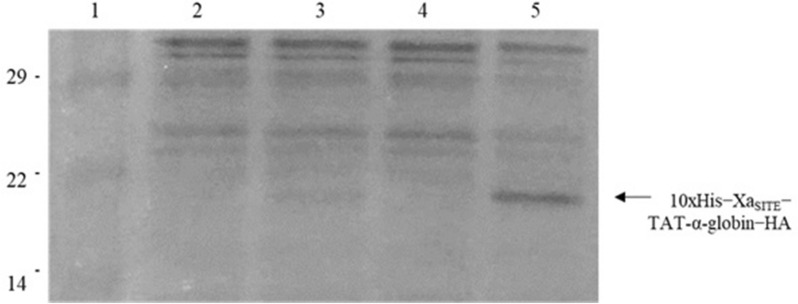
Cloning of the α-globin coding sequence, fused to the nucleotide sequence of TAT peptide was conducted and the human recombinant fusion proteins, 10xHis-Xa-α-globin-HA and 10xHis-Xa-TAT-α-globin-HA were produced. The ability of human recombinant 10xHis-Xa-α-globin-HA to interact in vitro with the previously produced 10xHis-Xa-TAT-β-globin-HA and form α-/β-globin heterodimers, was assessed and confirmed by size exclusion chromatography. The recombinant 10xHis-Xa-TAT-α-globin-HA was successfully delivered into human proerythroid K-562 cells, during the preliminary transduction evaluation experiments. Finally, the recombinant, TAT-fused α-globin was successfully transduced into RBCs, derived from HbH patients and reduced the formation of HbH-Inclusion Bodies, known to contain harmful β-globin chain tetramers.
Our data confirm the successful ex vivo transduction of recombinant α-globin chains in HbH RBCs to replace the missing a-globin chain and reduce the HbH-inclusion bodies, seen in α-thalassemias. These findings broaden the possibility of applying a Protein Replacement Therapy approach to module sever forms of α-thalassemia, using recombinant α-globin chains, through PTD technology.
α地中海贫血是一种先天性血红蛋白病,在严重形式的α地中海贫血(血红蛋白H病/巴氏胎儿血红蛋白病)中,其特征是α珠蛋白链缺乏和/或水平降低。本研究工作采用蛋白质替代疗法,以处理由血红蛋白H红细胞中β珠蛋白链过量引起的α地中海贫血表现。主要目标是生产与蛋白质转导结构域TAT融合的重组人α珠蛋白链,将其体外递送至血红蛋白H病患者的红细胞中,以替代内源性缺失的α珠蛋白链。
进行了与TAT肽核苷酸序列融合的α珠蛋白编码序列的克隆,并生产了人重组融合蛋白10xHis-Xa-α珠蛋白-HA和10xHis-Xa-TAT-α珠蛋白-HA。通过尺寸排阻色谱法评估并确认了人重组10xHis-Xa-α珠蛋白-HA与先前生产的10xHis-Xa-TAT-β珠蛋白-HA在体外相互作用并形成α/β珠蛋白异二聚体的能力。在初步转导评估实验中,重组10xHis-Xa-TAT-α珠蛋白-HA成功递送至人早幼红细胞K-562细胞中。最后,与TAT融合的重组α珠蛋白成功转导至来自血红蛋白H病患者的红细胞中,并减少了已知含有有害β珠蛋白链四聚体的血红蛋白H包涵体的形成。
我们的数据证实了重组α珠蛋白链在血红蛋白H红细胞中的体外成功转导,以替代缺失的α珠蛋白链并减少在α地中海贫血中所见的血红蛋白H包涵体。这些发现拓宽了通过PTD技术应用蛋白质替代疗法,使用重组α珠蛋白链来调节严重形式的α地中海贫血的可能性。