Departments of Genetics and Psychiatry, Washington University in St. Louis, St. Louis, MO, USA.
Transl Psychiatry. 2021 Jul 22;11(1):403. doi: 10.1038/s41398-021-01493-6.
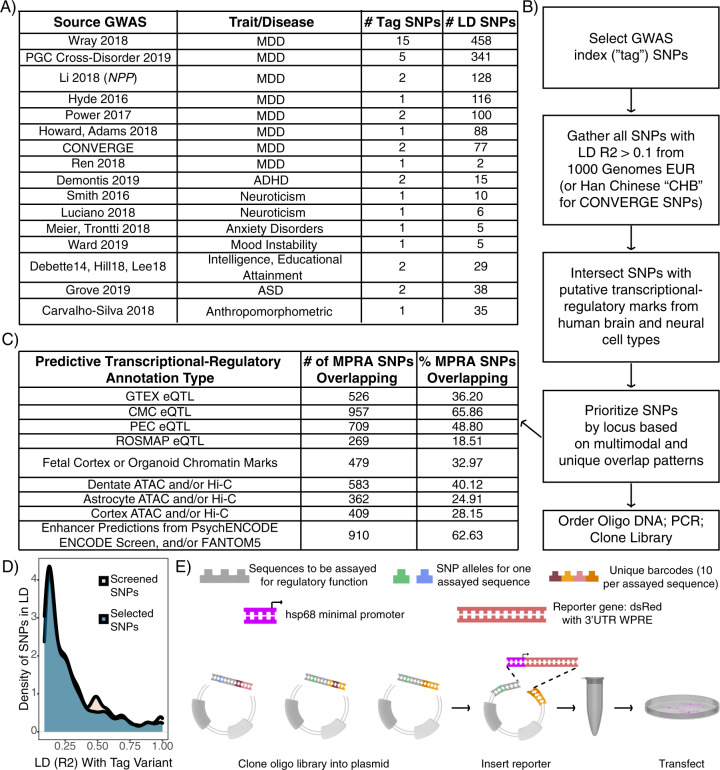
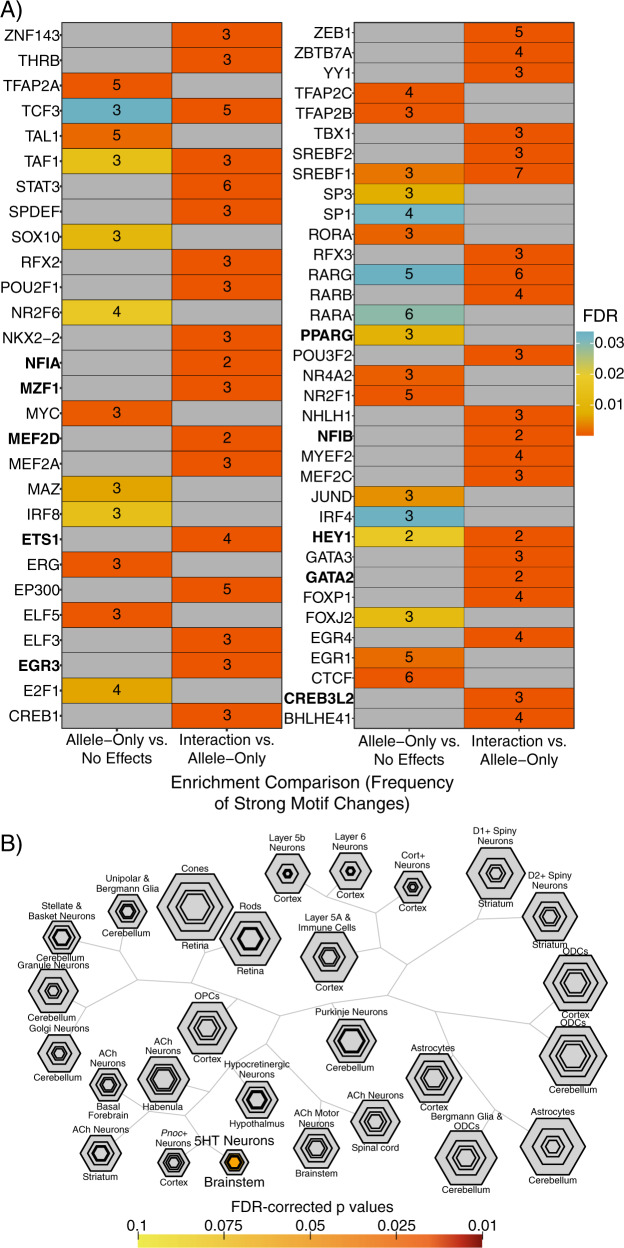
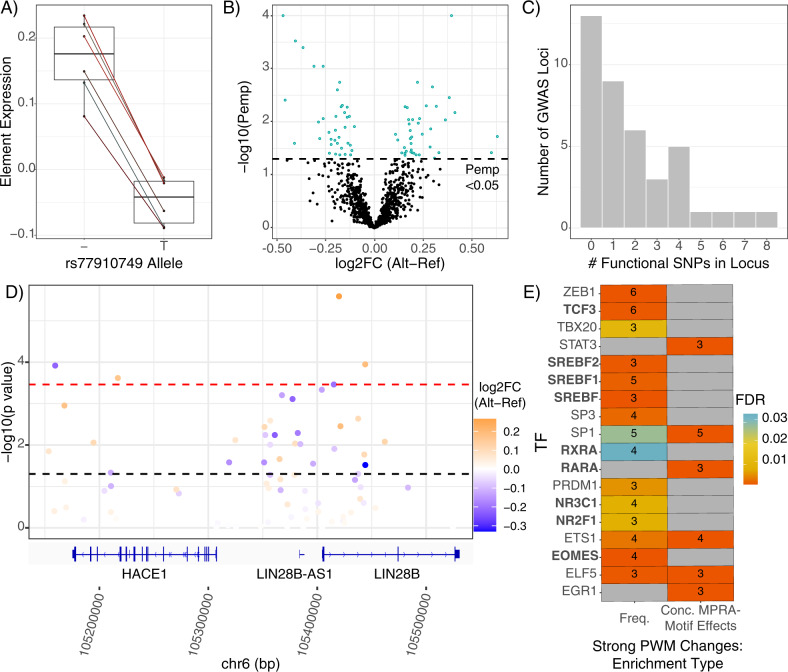
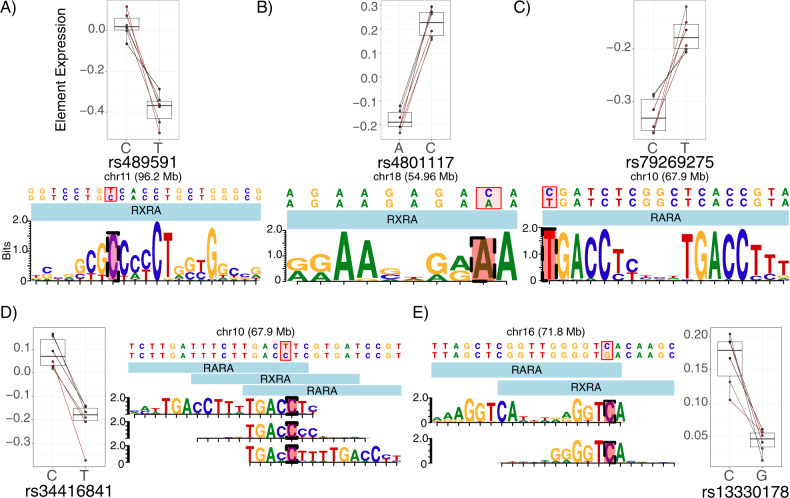
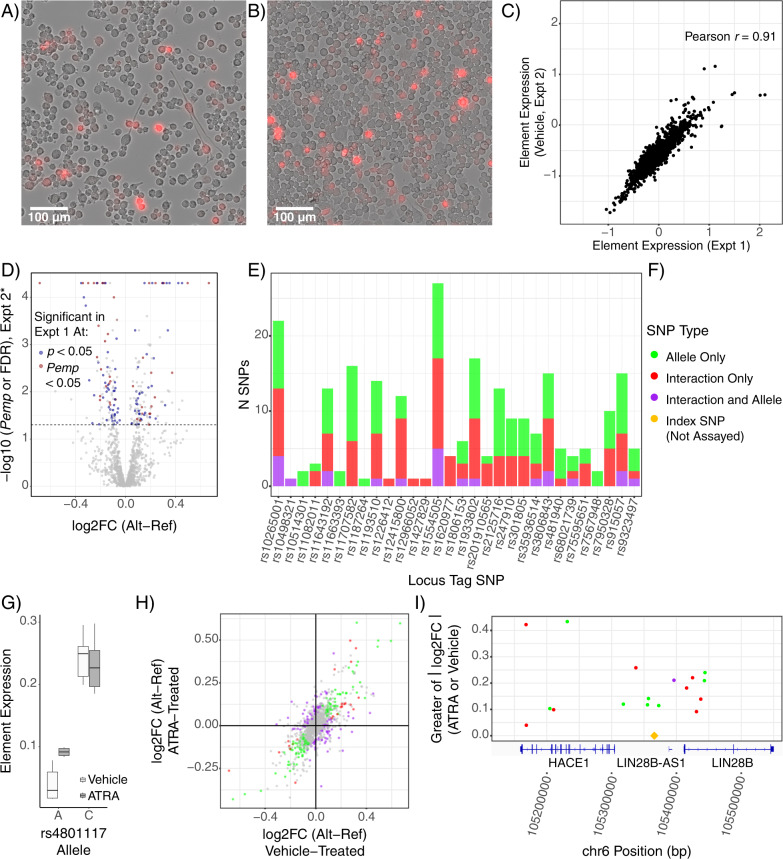
Family and population studies indicate clear heritability of major depressive disorder (MDD), though its underlying biology remains unclear. The majority of single-nucleotide polymorphism (SNP) linkage blocks associated with MDD by genome-wide association studies (GWASes) are believed to alter transcriptional regulators (e.g., enhancers, promoters) based on enrichment of marks correlated with these functions. A key to understanding MDD pathophysiology will be elucidation of which SNPs are functional and how such functional variants biologically converge to elicit the disease. Furthermore, retinoids can elicit MDD in patients and promote depressive-like behaviors in rodent models, acting via a regulatory system of retinoid receptor transcription factors (TFs). We therefore sought to simultaneously identify functional genetic variants and assess retinoid pathway regulation of MDD risk loci. Using Massively Parallel Reporter Assays (MPRAs), we functionally screened over 1000 SNPs prioritized from 39 neuropsychiatric trait/disease GWAS loci, selecting SNPs based on overlap with predicted regulatory features-including expression quantitative trait loci (eQTL) and histone marks-from human brains and cell cultures. We identified >100 SNPs with allelic effects on expression in a retinoid-responsive model system. Functional SNPs were enriched for binding sequences of retinoic acid-receptive transcription factors (TFs), with additional allelic differences unmasked by treatment with all-trans retinoic acid (ATRA). Finally, motifs overrepresented across functional SNPs corresponded to TFs highly specific to serotonergic neurons, suggesting an in vivo site of action. Our application of MPRAs to screen MDD-associated SNPs suggests a shared transcriptional-regulatory program across loci, a component of which is unmasked by retinoids.
家族和人口研究表明,重度抑郁症(MDD)具有明显的遗传性,但其潜在生物学机制仍不清楚。全基因组关联研究(GWAS)中与 MDD 相关的大多数单核苷酸多态性(SNP)连锁块,据信是基于与这些功能相关的标记富集,改变转录调节剂(如增强子、启动子)。理解 MDD 病理生理学的关键将是阐明哪些 SNP 是功能性的,以及这些功能性变体如何通过生物方式汇聚以引发疾病。此外,视黄酸可以在患者中引发 MDD ,并在啮齿动物模型中促进抑郁样行为,通过视黄酸受体转录因子(TFs)的调节系统发挥作用。因此,我们试图同时鉴定功能性遗传变异,并评估视黄酸通路对 MDD 风险位点的调控作用。我们使用大规模平行报告基因检测(MPRAs),从 39 个神经精神特征/疾病 GWAS 位点中筛选了超过 1000 个优先的 SNP,根据与人类大脑和细胞培养物中预测的调节特征(包括表达数量性状基因座(eQTL)和组蛋白标记)的重叠,选择了 SNP。我们在一个视黄酸反应性模型系统中鉴定了 >100 个具有等位基因表达效应的 SNP。功能 SNP 富含视黄酸受体转录因子(TFs)的结合序列,用全反式视黄酸(ATRA)处理后,揭示了更多的等位基因差异。功能 SNP 上过度表达的基序对应于高度特异于 5-羟色胺能神经元的 TF,提示存在体内作用部位。我们应用 MPRAs 筛选与 MDD 相关的 SNP 表明,多个位点之间存在共享的转录调节程序,其中一部分是由视黄酸揭示的。