Laboratoire de Chimie Organique Hétérocyclique, URAC 21, Pôle de Compétences Pharmaco-Chimie, Faculté Des Sciences, Université Mohammed V, BP 10014, Rabat, Morocco.
Department of Theoretical and Applied Chemistry, South Ural State University, Lenin prospect 76, Chelyabinsk, 454080, Russian Federation.
Appl Biochem Biotechnol. 2021 Nov;193(11):3602-3623. doi: 10.1007/s12010-021-03615-8. Epub 2021 Jul 29.
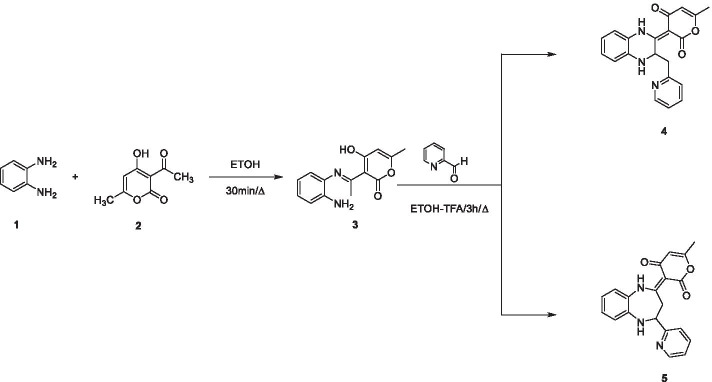
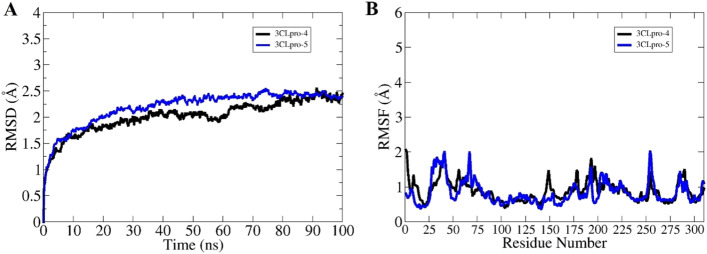
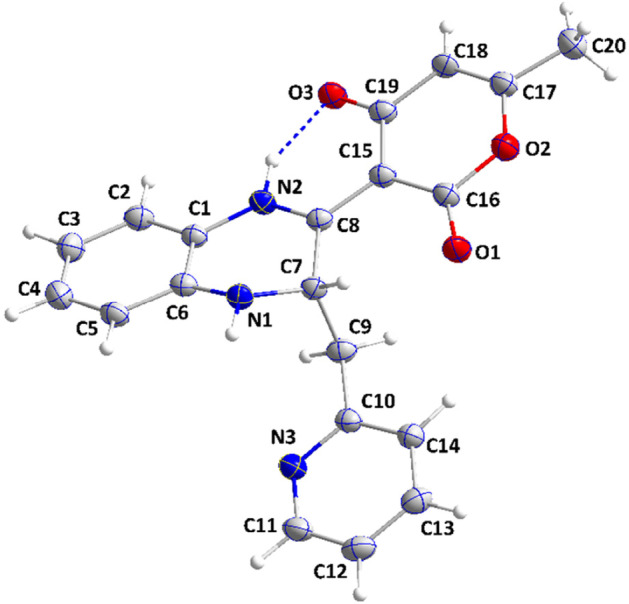
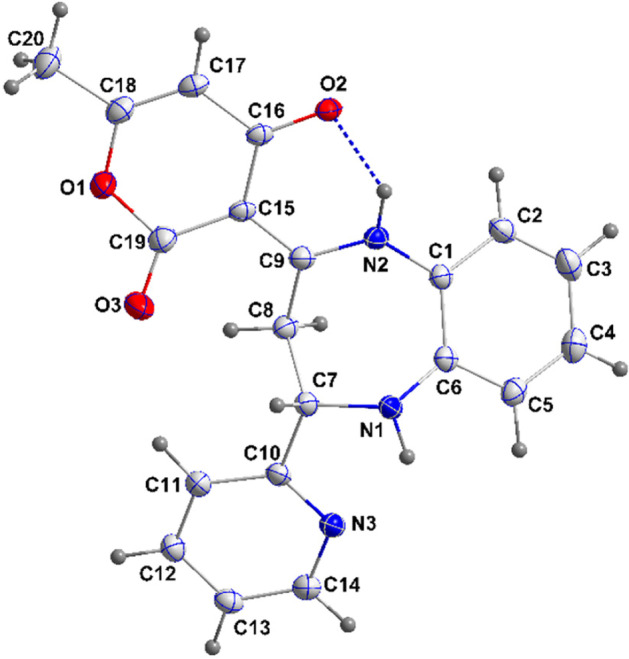
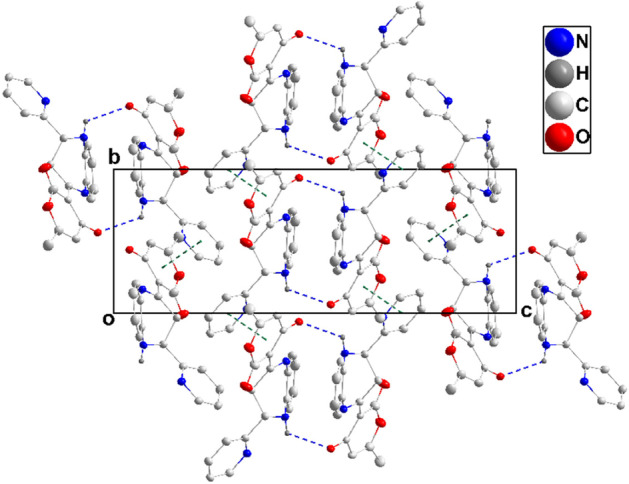
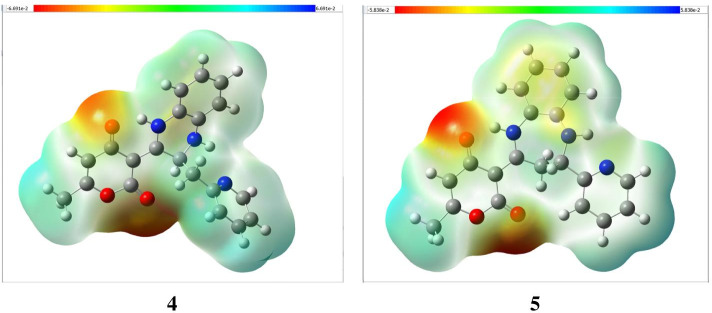
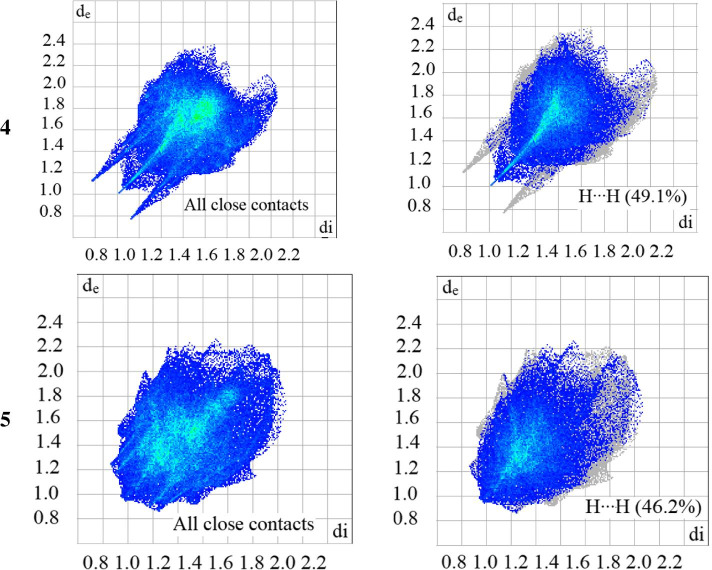
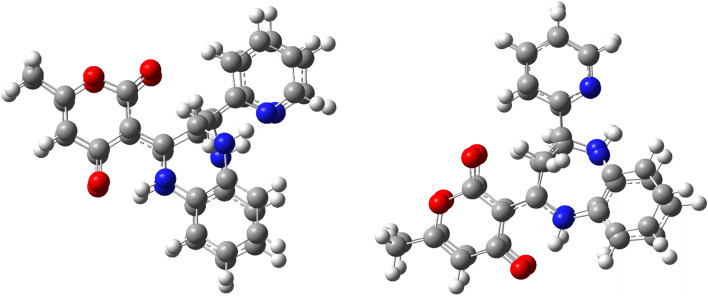
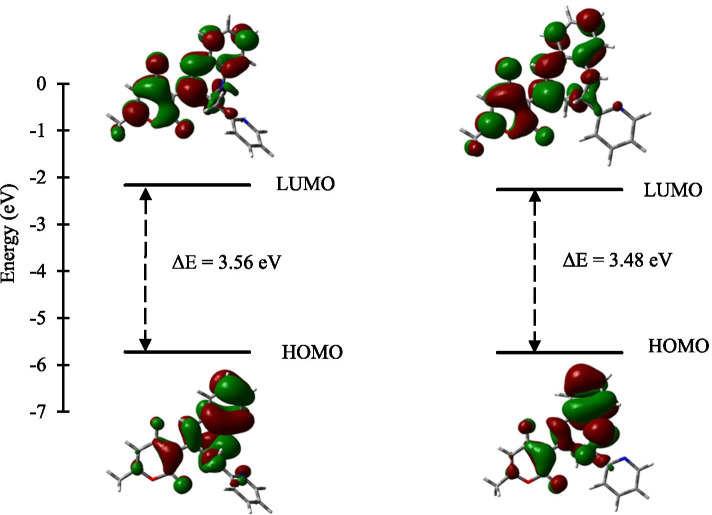
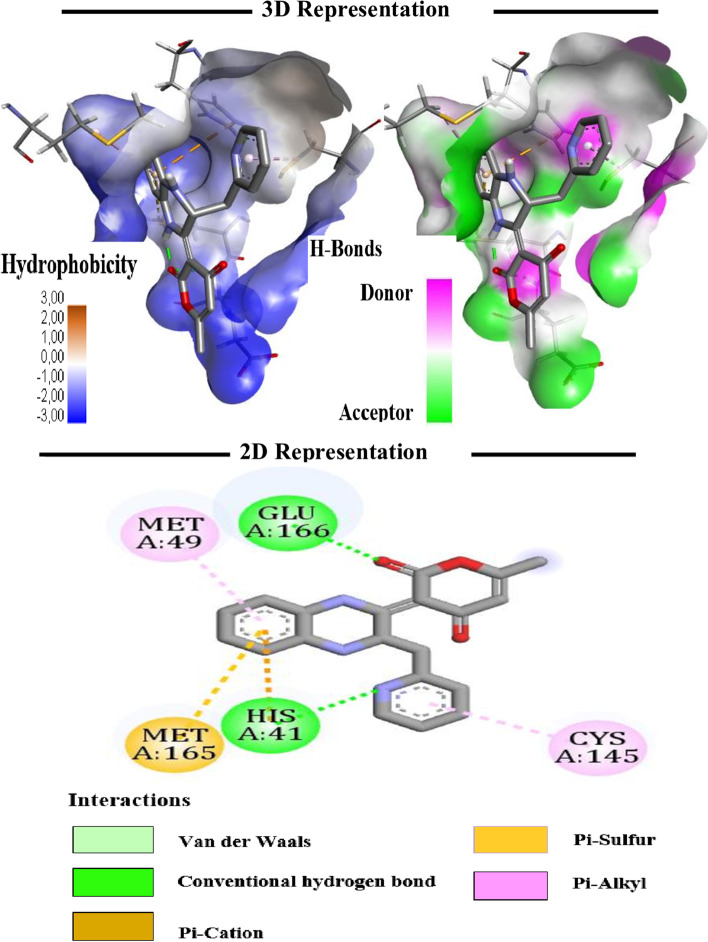
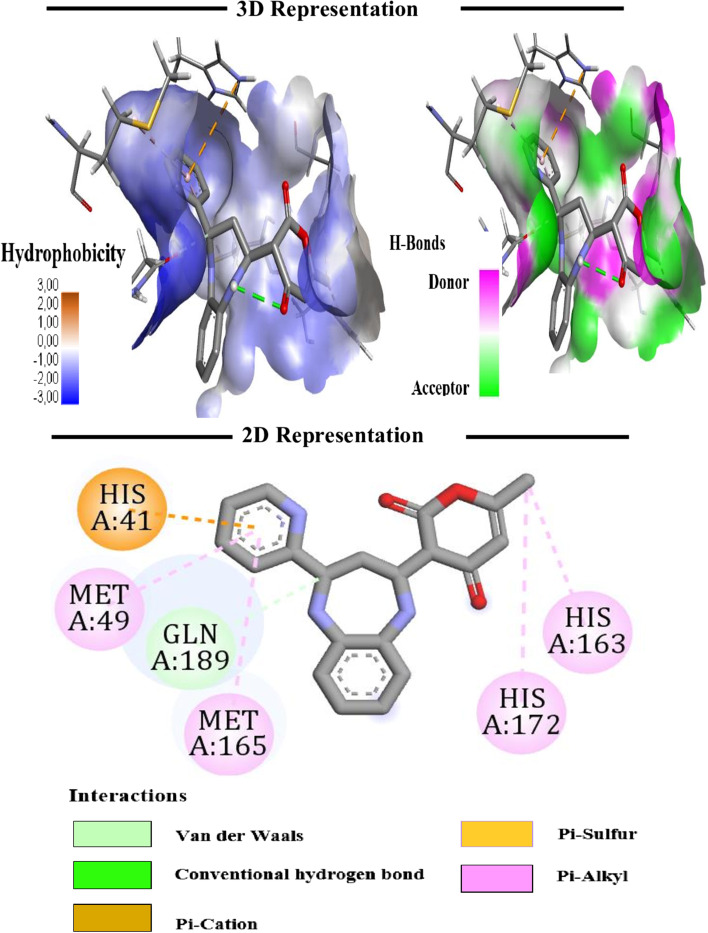
The novel coronavirus disease that arises in the end of 2019 (COVID-19) in Wuhan, China, has rapidly spread over the globe and was considered as a world pandemic. Currently, various antiviral therapies or vaccines are available, and many researches are ongoing for further treatments. Targeting the coronavirus' main protease (key enzyme: 3CLpro) is growing in importance in anti-SARS-CoV-2 drug discovery process. The present study aims at predicting the antiviral activity of two novel compounds using in silico approaches that might become potential leads against SARS-CoV-2. The 3D structures of the new compounds are elucidated by single-crystal X-ray techniques. The interactions between different units of 4 and 5 were emphasized by analyzing their corresponding Hirshfeld surfaces and ESP plots. NBO and FMO analyses were investigated as well. Molecular docking combined with molecular dynamics simulations (MDs) was performed to investigate the binding modes and molecular interactions of 4 and 5 with the amino acids of coronavirus main protease (6LU7) protein. The best docking scores were obtained for both ligands through the major binding interactions via hydrogen/hydrophobic bonds with the key amino acids in the active site: HIS41, CYS145, MET49, MET165, HIS172, and GLU166 amino acids. A MD simulation study was also performed for 100 ns to validate the stability behavior of the main protease 3CLpro-ligand complexes. The MD simulation study successfully confirmed the stability of the ligands in the binding site as potent anti-SARS-CoV-2 (COVID-19) inhibitors. Additionally, MMPBSA energy of both docked complexes was determined as a validation assay of docking and MD simulations to validate compound conformation and interaction stability with 3CLpro. The synthesized compounds might be helpful in the fight against COVID-19 prior to biological activity confirmation in vitro and in vivo.
2019 年底在中国武汉出现的新型冠状病毒病(COVID-19)迅速在全球蔓延,并被认为是一种世界大流行。目前,有多种抗病毒疗法或疫苗可用,许多研究正在进行中以寻求进一步的治疗方法。针对冠状病毒的主要蛋白酶(关键酶:3CLpro)的靶向治疗在抗 SARS-CoV-2 药物发现过程中变得越来越重要。本研究旨在通过计算机模拟方法预测两种新型化合物的抗病毒活性,这些化合物可能成为针对 SARS-CoV-2 的潜在先导化合物。通过单晶 X 射线技术阐明了新化合物的 3D 结构。通过分析相应的 Hirshfeld 表面和 ESP 图强调了 4 和 5 不同单元之间的相互作用。还研究了 NBO 和 FMO 分析。进行了分子对接结合分子动力学模拟(MDs),以研究 4 和 5 与冠状病毒主要蛋白酶(6LU7)蛋白的氨基酸的结合模式和分子相互作用。通过主要的氢键/疏水性结合与活性位点中的关键氨基酸:HIS41、CYS145、MET49、MET165、HIS172 和 GLU166 氨基酸,两种配体都获得了最佳的对接得分。还进行了 100ns 的 MD 模拟研究,以验证主要蛋白酶 3CLpro-配体复合物的稳定性行为。MD 模拟研究成功地证实了配体在结合位点中的稳定性,作为有效的抗 SARS-CoV-2(COVID-19)抑制剂。此外,还确定了两个对接复合物的 MMPBSA 能量,作为对接和 MD 模拟的验证试验,以验证化合物构象和与 3CLpro 的相互作用稳定性。在体外和体内生物活性确认之前,合成的化合物可能有助于对抗 COVID-19。