Boukerb Amine M, Noël Cyril, Quenot Emmanuelle, Cadiou Bernard, Chevé Julien, Quintric Laure, Cormier Alexandre, Dantan Luc, Gourmelon Michèle
IFREMER, RBE-SGMM-LSEM, Laboratoire Santé Environnement Microbiologie, Plouzané, France.
IFREMER - PDG-IRSI-SEBIMER, Plouzané, France.
Front Microbiol. 2021 Jul 14;12:697553. doi: 10.3389/fmicb.2021.697553. eCollection 2021.
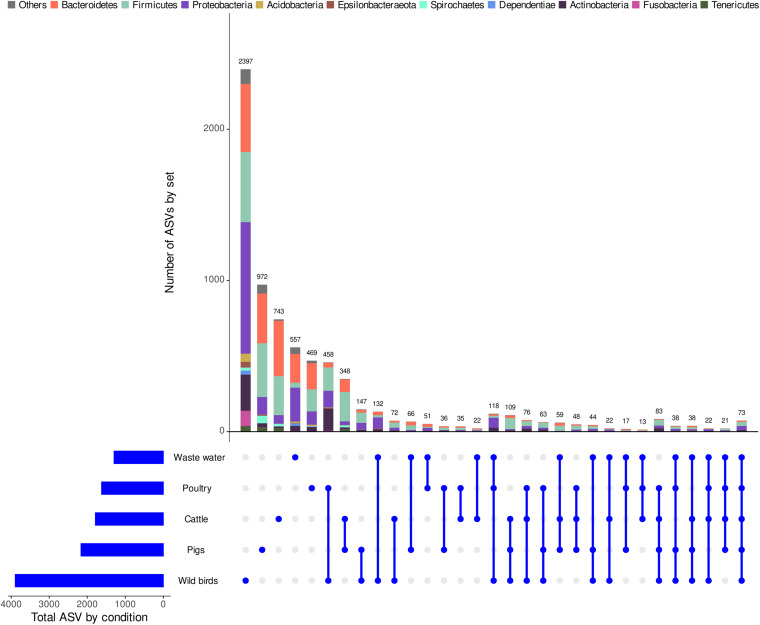
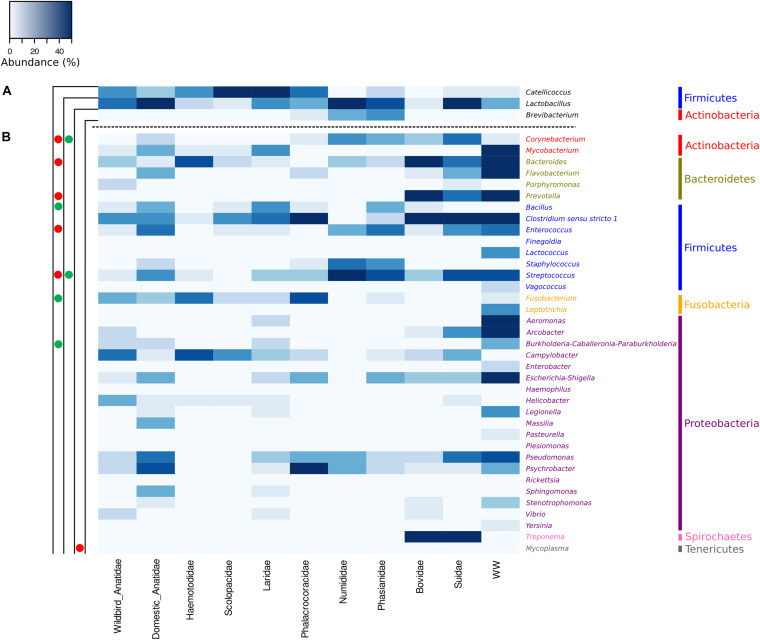
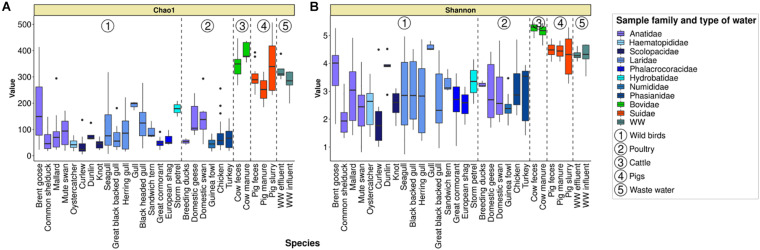
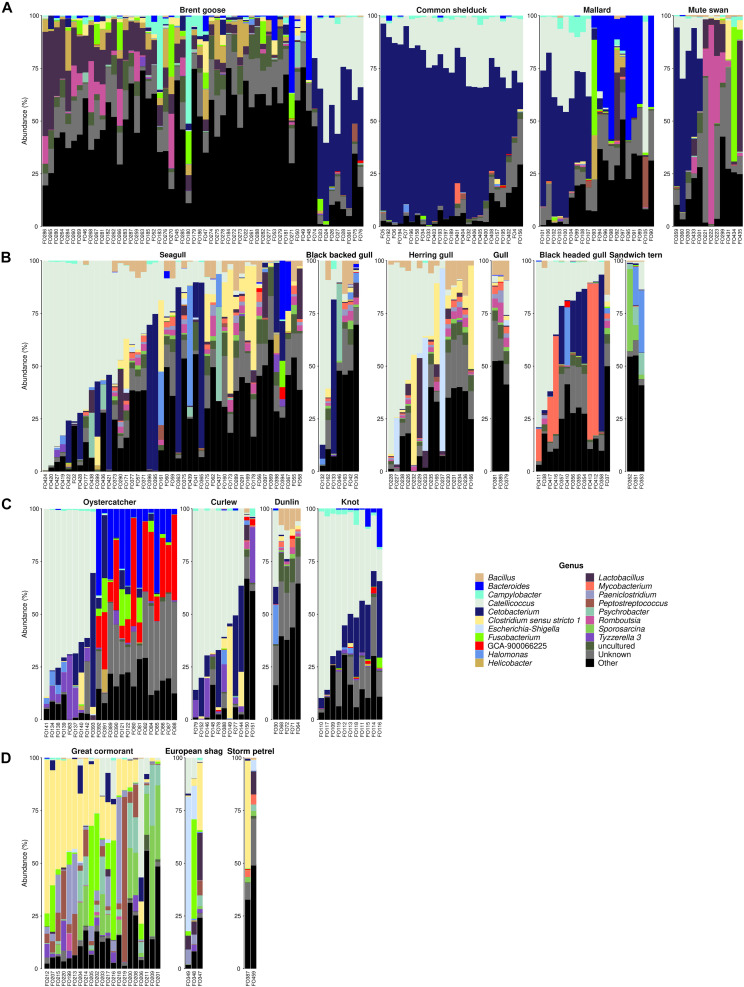
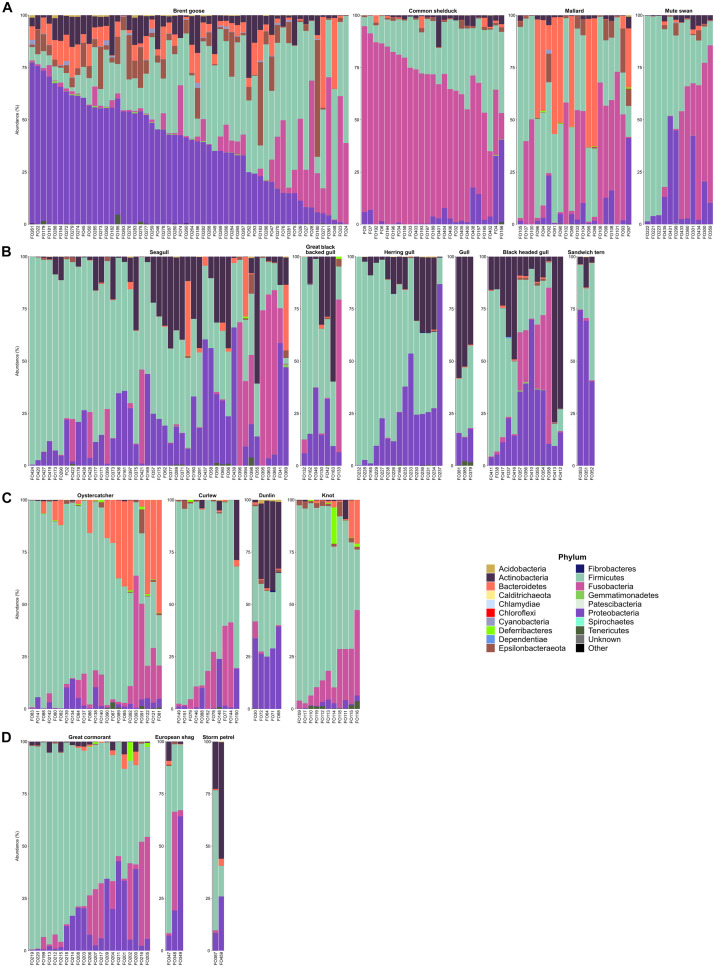
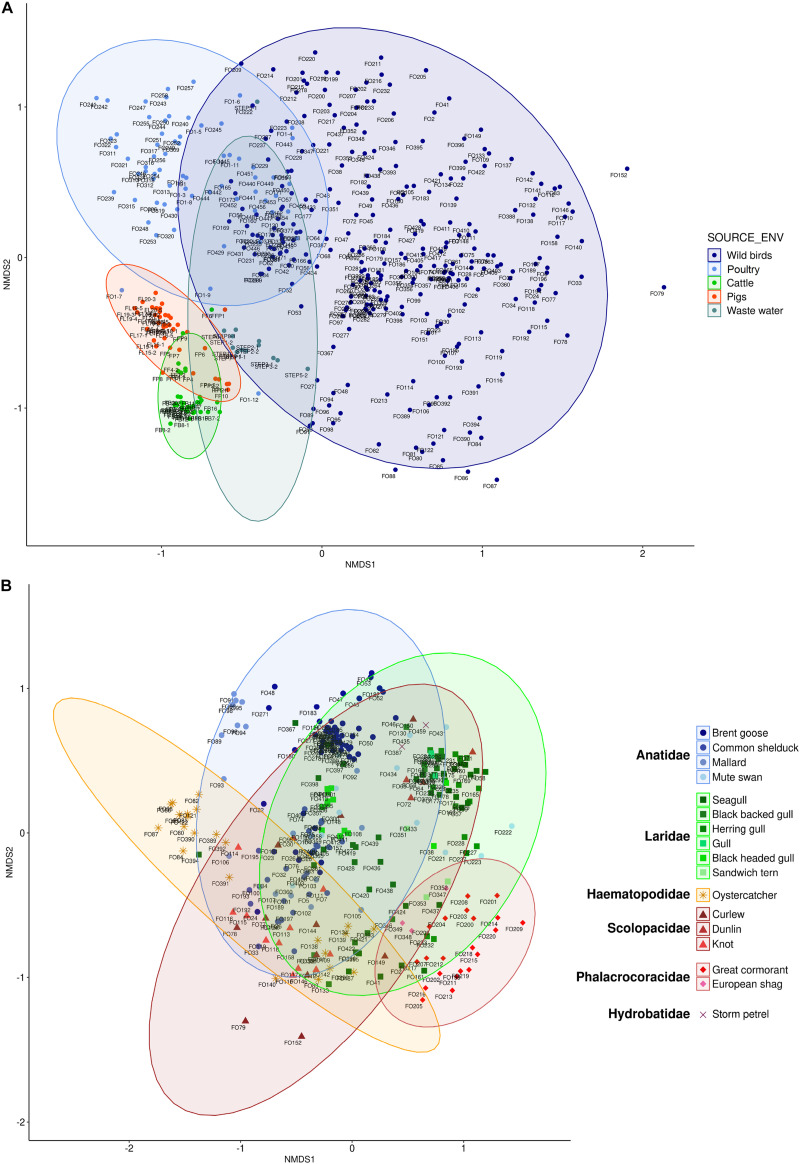
Fecal pollution in coastal areas is of a high concern since it affects bathing and shellfish harvesting activities. Wild waterbirds are non-negligible in the overall signal of the detectable pollution. Yet, studies on wild waterbirds' gut microbiota focus on migratory trajectories and feeding impact on their shape, rare studies address their comparison to other sources and develop quantitative PCR (qPCR)-based Microbial Source Tracking (MST) markers to detect such pollution. Thus, by using 16S rRNA amplicon high-throughput sequencing, the aims of this study were (i) to explore and compare fecal bacterial communities from wild waterbirds (i.e., six families and 15 species, = 275 samples) to that of poultry, cattle, pigs, and influent/effluent of wastewater treatment plants ( = 150 samples) and (ii) to develop new MST markers for waterbirds. Significant differences were observed between wild waterbirds and the four other groups. We identified 7,349 Amplicon Sequence Variants (ASVs) from the hypervariable V3-V4 region. Firmicutes and Proteobacteria and, in a lesser extent, Actinobacteria and Bacteroidetes were ubiquitous while Fusobacteria and Epsilonbacteraeota were mainly present in wild waterbirds. The clustering of samples in non-metric multidimensional scaling (NMDS) ordination indicated a by-group clustering shape, with a high diversity within wild waterbirds. In addition, the structure of the bacterial communities was distinct according to bird and/or animal species and families (Adonis = 0.13, = 10, Adonis = 0.11, = 10, respectively). The Analysis of Composition of Microbiomes (ANCOM) showed that the wild waterbird group differed from the others by the significant presence of sequences from Fusobacteriaceae ( = 566) and Enterococcaceae ( = 565) families, corresponding to the ( = 1427) and ( = 1427) genera, respectively. Altogether, our results suggest that some waterbird members present distinct fecal microbiomes allowing the design of qPCR MST markers. For instance, a swan- and an oystercatcher-associated markers (named Swan_2 and Oyscab, respectively) have been developed. Moreover, bacterial genera harboring potential human pathogens associated to bird droppings were detected in our dataset, including enteric pathogens, i.e., , , , and , and environmental pathogens, i.e., and . Future studies involving other wildlife hosts may improve gut microbiome studies and MST marker development, helping mitigation of yet unknown fecal pollution sources.
沿海地区的粪便污染备受关注,因为它会影响游泳和贝类捕捞活动。在可检测到的污染总体信号中,野生水鸟的影响不可忽视。然而,关于野生水鸟肠道微生物群的研究主要集中在迁徙轨迹及其觅食对其体型的影响,很少有研究将它们与其他污染源进行比较,也很少开发基于定量聚合酶链反应(qPCR)的微生物源追踪(MST)标记来检测此类污染。因此,本研究通过16S rRNA扩增子高通量测序,旨在(i)探索并比较野生水鸟(即6个科15个物种,n = 275个样本)与家禽、牛、猪以及污水处理厂进水/出水(n = 150个样本)的粪便细菌群落,(ii)开发针对水鸟的新型MST标记。野生水鸟与其他四组之间存在显著差异。我们从高变V3 - V4区域鉴定出7349个扩增子序列变体(ASV)。厚壁菌门和变形菌门,以及在较小程度上的放线菌门和拟杆菌门普遍存在,而梭杆菌门和ε - 变形菌门主要存在于野生水鸟中。非度量多维尺度分析(NMDS)排序中的样本聚类显示出按组聚类的形态,野生水鸟内部具有高度多样性。此外,细菌群落结构因鸟类和/或动物的物种及科而异(Adonis检验,R2 = 0.13,p = 10,Adonis检验,R2 = 0.11,p = 10)。微生物群落组成分析(ANCOM)表明,野生水鸟组与其他组的不同之处在于显著存在来自梭杆菌科(n = 566)和肠球菌科(n = 565)的序列,分别对应于梭杆菌属(n = 1427)和肠球菌属(n = 1427)。总之,我们的结果表明,一些水鸟成员具有独特的粪便微生物群,这使得设计qPCR MST标记成为可能。例如,已经开发出了与天鹅和蛎鹬相关的标记(分别命名为Swan_2和Oyscab)。此外,在我们的数据集中检测到了与鸟粪相关的携带潜在人类病原体的细菌属,包括肠道病原体,即大肠杆菌、沙门氏菌、志贺氏菌和弯曲杆菌,以及环境病原体,即气单胞菌和假单胞菌。未来涉及其他野生动物宿主的研究可能会改善肠道微生物群研究和MST标记开发,有助于减轻尚未明确的粪便污染源。