Schwob Guillaume, Segovia Nicolás I, González-Wevar Claudio, Cabrol Léa, Orlando Julieta, Poulin Elie
Departamento de Ciencias Ecológicas, Facultad de Ciencias, Universidad de Chile, Santiago, Chile.
Instituto de Ecología y Biodiversidad, Santiago, Chile.
Front Microbiol. 2021 Jul 14;12:703792. doi: 10.3389/fmicb.2021.703792. eCollection 2021.
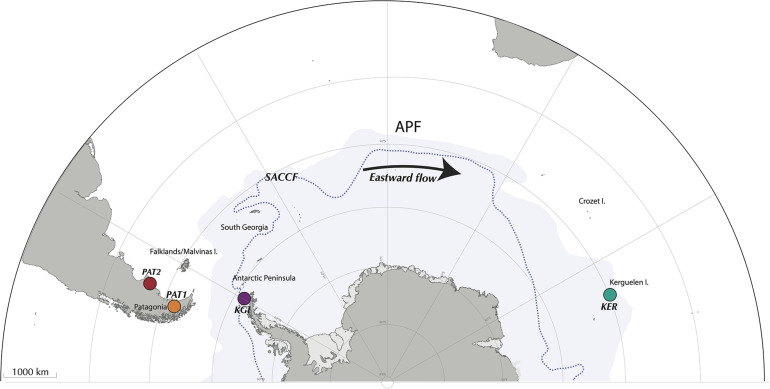
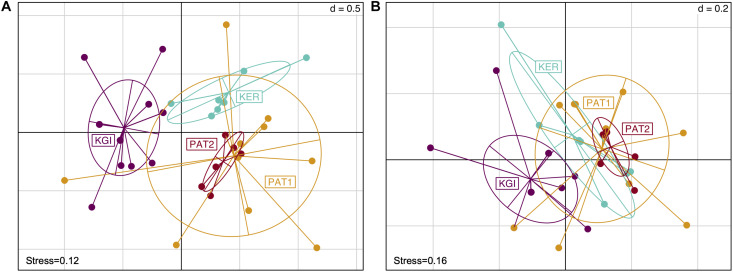
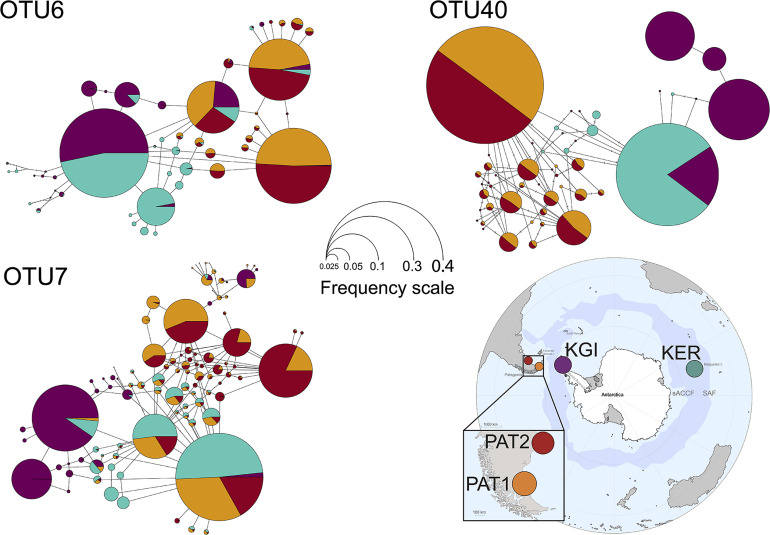
Most of the microbial biogeographic patterns in the oceans have been depicted at the whole community level, leaving out finer taxonomic resolution (i.e., microdiversity) that is crucial to conduct intra-population phylogeographic study, as commonly done for macroorganisms. Here, we present a new approach to unravel the bacterial phylogeographic patterns combining community-wide survey by 16S rRNA gene metabarcoding and intra-species resolution through the oligotyping method, allowing robust estimations of genetic and phylogeographic indices, and migration parameters. As a proof-of-concept, we focused on the bacterial genus across three distant biogeographic provinces of the Southern Ocean; maritime Antarctica, sub-Antarctic Islands, and Patagonia. Each targeted operational taxonomic units were characterized by a substantial intrapopulation microdiversity, and significant genetic differentiation and phylogeographic structure among the three provinces. Gene flow estimations among populations support the role of the Antarctic Polar Front as a biogeographic barrier to bacterial dispersal between Antarctic and sub-Antarctic provinces. Conversely, the Antarctic Circumpolar Current appears as the main driver of gene flow, connecting sub-Antarctic Islands with Patagonia and maritime Antarctica. Additionally, historical processes (drift and dispersal limitation) govern up to 86% of the spatial turnover among populations. Overall, our approach bridges the gap between microbial and macrobial ecology by revealing strong congruency with macroorganisms distribution patterns at the populational level, shaped by the same oceanographic structures and ecological processes.
海洋中大多数微生物生物地理模式都是在整个群落水平上描绘的,忽略了更精细的分类分辨率(即微观多样性),而这种分辨率对于像研究大型生物那样进行种群内系统地理学研究至关重要。在此,我们提出一种新方法,将通过16S rRNA基因代谢条形码进行的群落范围调查与通过寡核苷酸分型方法进行的种内分辨率相结合,以揭示细菌生物地理模式,从而能够可靠地估计遗传和生物地理指数以及迁移参数。作为概念验证,我们聚焦于南大洋三个遥远生物地理省份中的细菌属;南极海洋、亚南极岛屿和巴塔哥尼亚。每个目标操作分类单元都具有大量的种群内微观多样性,并且这三个省份之间存在显著的遗传分化和生物地理结构。种群间的基因流估计支持南极极锋作为南极和亚南极省份之间细菌扩散的生物地理屏障的作用。相反,南极绕极流似乎是基因流的主要驱动力,将亚南极岛屿与巴塔哥尼亚和南极海洋连接起来。此外,历史过程(漂移和扩散限制)控制了种群间高达86%的空间周转率。总体而言,我们的方法通过揭示在种群水平上与大型生物分布模式的强烈一致性,弥合了微生物生态学和大型生物生态学之间的差距,这种一致性是由相同的海洋学结构和生态过程塑造的。