Chen Dian, Wu Wenliang, Yi Lingling, Feng Yuchen, Chang Chenli, Chen Shengchong, Gao Jiali, Chen Gongqi, Zhen Guohua
Division of Respiratory and Critical Care Medicine, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Key Laboratory of Respiratory Diseases, National Health Commission of People's Republic of China, and National Clinical Research Center for Respiratory Diseases, Wuhan, China.
Front Mol Biosci. 2021 Jul 16;8:703307. doi: 10.3389/fmolb.2021.703307. eCollection 2021.
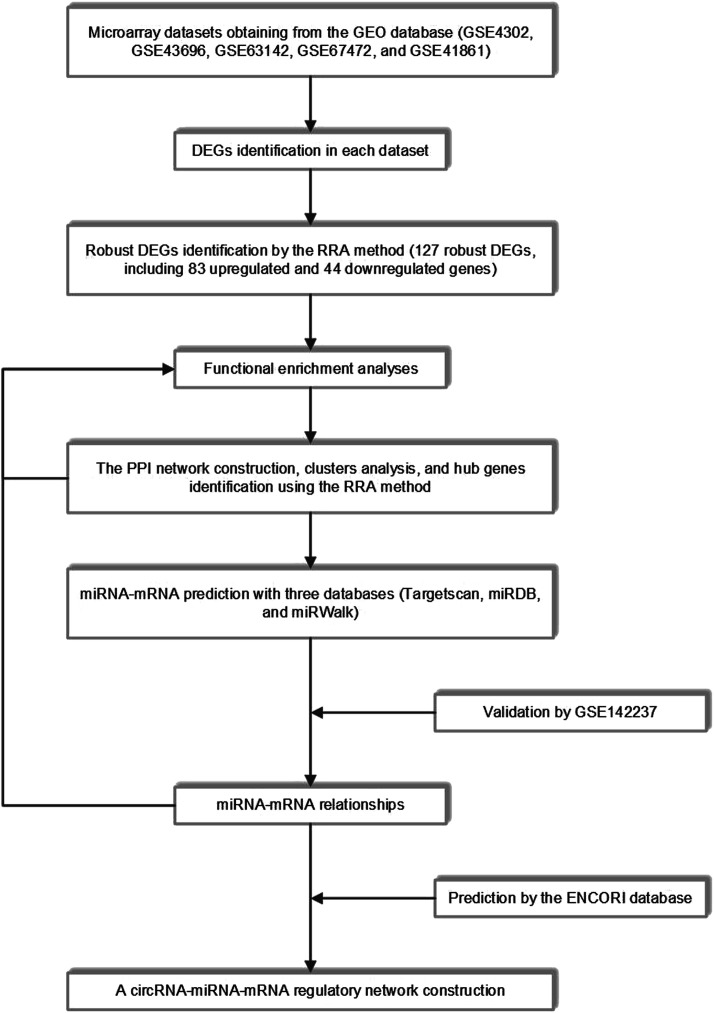
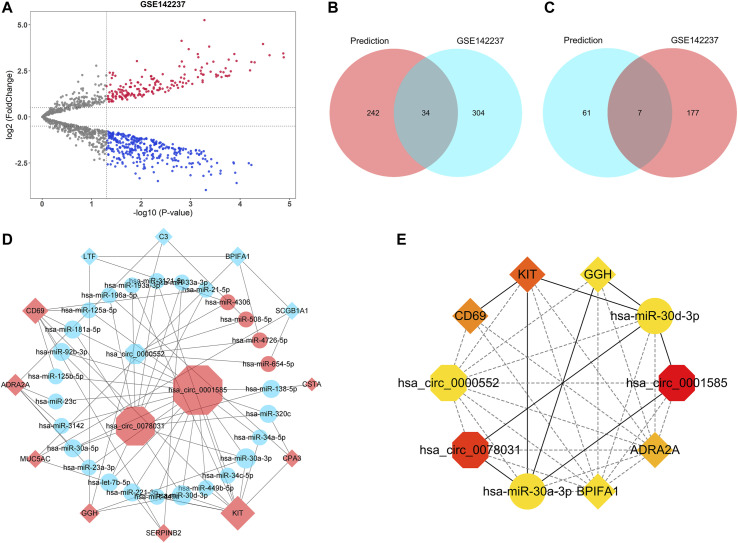
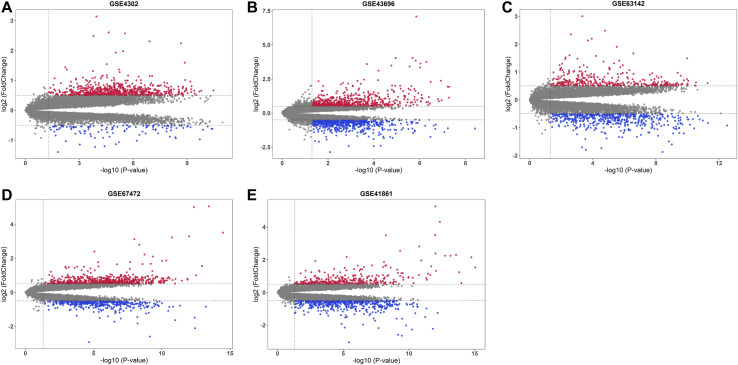
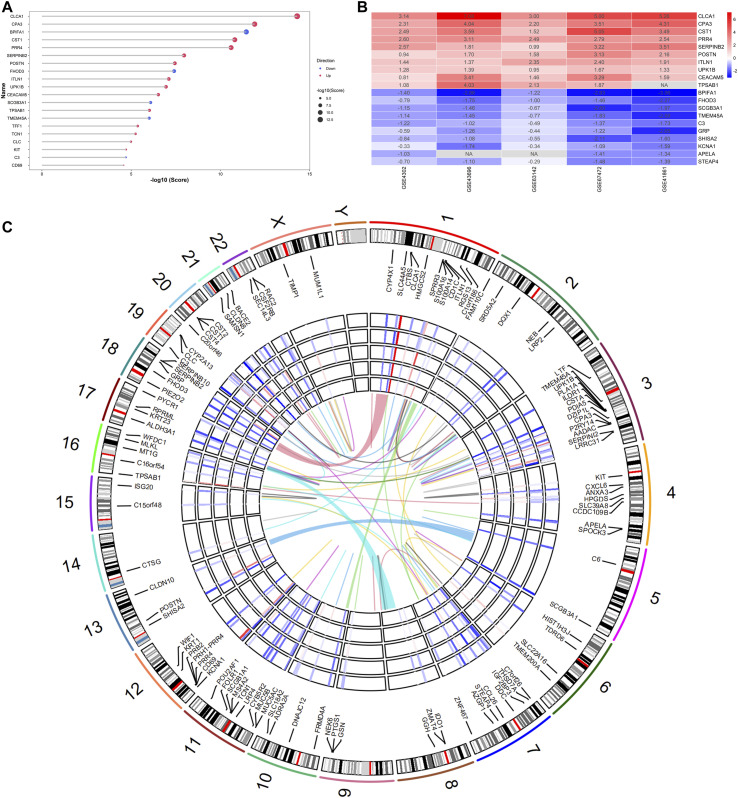
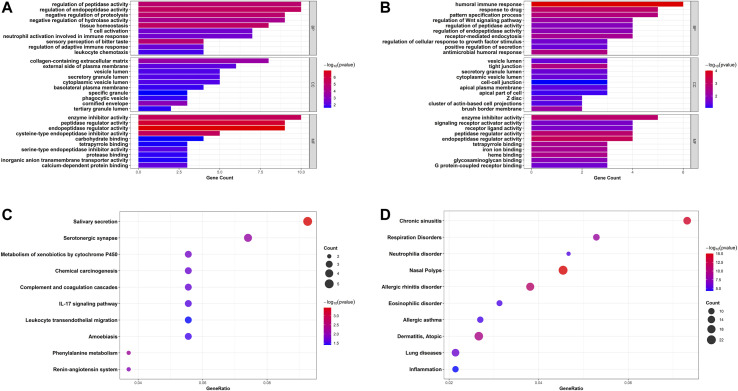
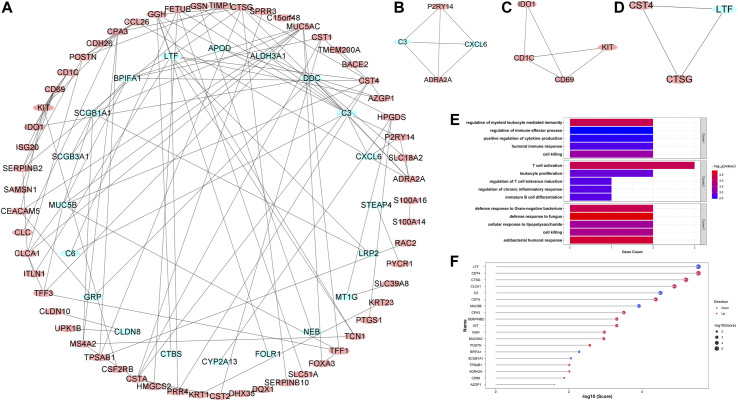
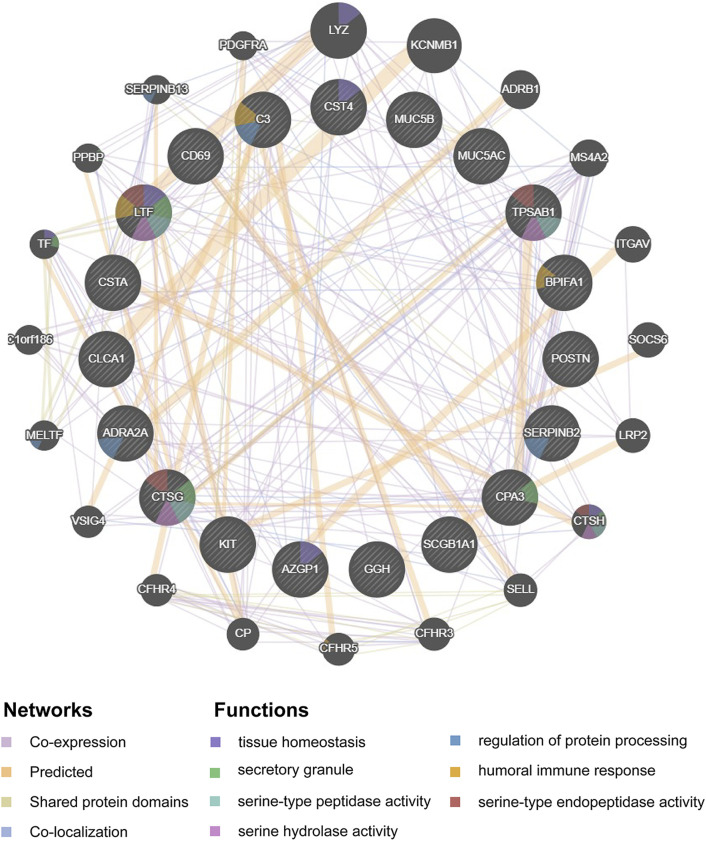
Asthma is one of the most prevalent chronic respiratory diseases worldwide. Bronchial epithelial cells play a critical role in the pathogenesis of asthma. Circular RNAs (circRNAs) act as microRNA (miRNA) sponges to regulate downstream gene expression. However, the role of epithelial circRNAs in asthma remains to be investigated. This study aims to explore the potential circRNA-miRNA-messenger RNA (mRNA) regulatory network in asthma by integrated analysis of publicly available microarray datasets. Five mRNA microarray datasets derived from bronchial brushing samples from asthma patients and control subjects were downloaded from the Gene Expression Omnibus (GEO) database. The robust rank aggregation (RRA) method was used to identify robust differentially expressed genes (DEGs) in bronchial epithelial cells between asthma patients and controls. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were used to annotate the functions of the DEGs. Protein-protein interaction (PPI) analysis was performed to identify hub genes. Three miRNA databases (Targetscan, miRDB, and miRWalk) were used to predict the miRNAs which potentially target the hub genes. A miRNA microarray dataset derived from bronchial brushings was used to validate the miRNA-mRNA relationships. Finally, a circRNA-miRNA-mRNA network was constructed via the ENCORI database. A total of 127 robust DEGs in bronchial epithelial cells between steroid-naïve asthma patients ( = 272) and healthy controls ( = 165) were identified from five mRNA microarray datasets. Enrichment analyses showed that DEGs were mainly enriched in several biological processes related to asthma, including humoral immune response, salivary secretion, and IL-17 signaling pathway. Nineteen hub genes were identified and were used to construct a potential epithelial circRNA-miRNA-mRNA network. The top 10 competing endogenous RNAs were , , , , , , , , , and . Our study reveals a potential role for epithelial circRNA-miRNA-mRNA network in the pathogenesis of asthma.
哮喘是全球最常见的慢性呼吸道疾病之一。支气管上皮细胞在哮喘发病机制中起关键作用。环状RNA(circRNAs)作为微小RNA(miRNA)海绵调节下游基因表达。然而,上皮circRNAs在哮喘中的作用仍有待研究。本研究旨在通过对公开可用的微阵列数据集进行综合分析,探索哮喘中潜在的circRNA-miRNA-信使RNA(mRNA)调控网络。从基因表达综合数据库(GEO)下载了五个来自哮喘患者和对照受试者支气管刷检样本的mRNA微阵列数据集。采用稳健秩聚合(RRA)方法鉴定哮喘患者和对照之间支气管上皮细胞中稳健的差异表达基因(DEGs)。基因本体(GO)和京都基因与基因组百科全书(KEGG)富集分析用于注释DEGs的功能。进行蛋白质-蛋白质相互作用(PPI)分析以鉴定枢纽基因。使用三个miRNA数据库(Targetscan、miRDB和miRWalk)预测可能靶向枢纽基因的miRNA。一个来自支气管刷检的miRNA微阵列数据集用于验证miRNA-mRNA关系。最后,通过ENCORI数据库构建circRNA-miRNA-mRNA网络。从五个mRNA微阵列数据集中鉴定出初治哮喘患者(n = 272)和健康对照(n = 165)之间支气管上皮细胞中共有127个稳健的DEGs。富集分析表明,DEGs主要富集在与哮喘相关的几个生物学过程中,包括体液免疫反应、唾液分泌和IL-17信号通路。鉴定出19个枢纽基因,并用于构建潜在的上皮circRNA-miRNA-mRNA网络。排名前十的竞争性内源性RNA分别是……(此处原文未完整列出具体基因名称)。我们的研究揭示了上皮circRNA-miRNA-mRNA网络在哮喘发病机制中的潜在作用。