Gong Xue, Huang Cheng, Yang Xun, Chen Jianjun, Pu Juncai, He Yong, Xie Peng
NHC Key Laboratory of Diagnosis and Treatment on Brain Functional Diseases, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
Department of Neurology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
Front Neurosci. 2021 Jul 19;15:701355. doi: 10.3389/fnins.2021.701355. eCollection 2021.
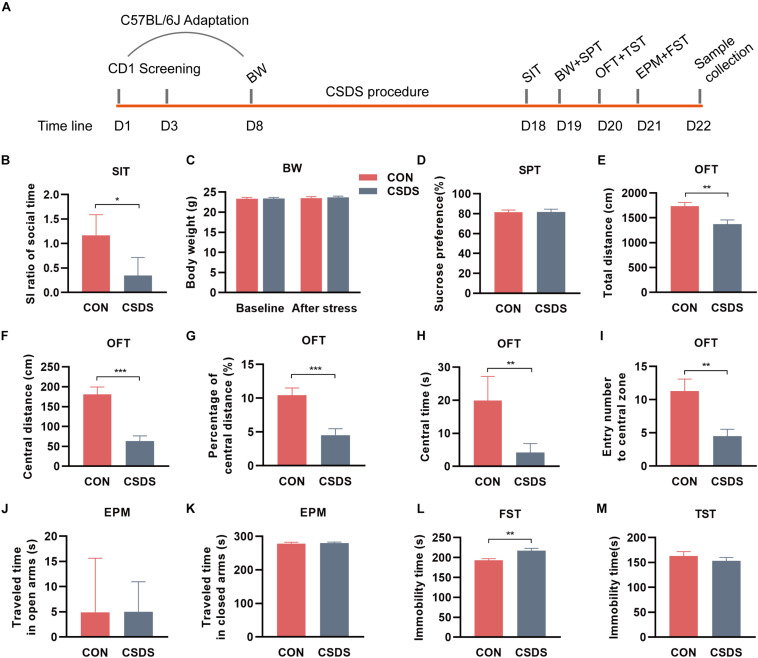
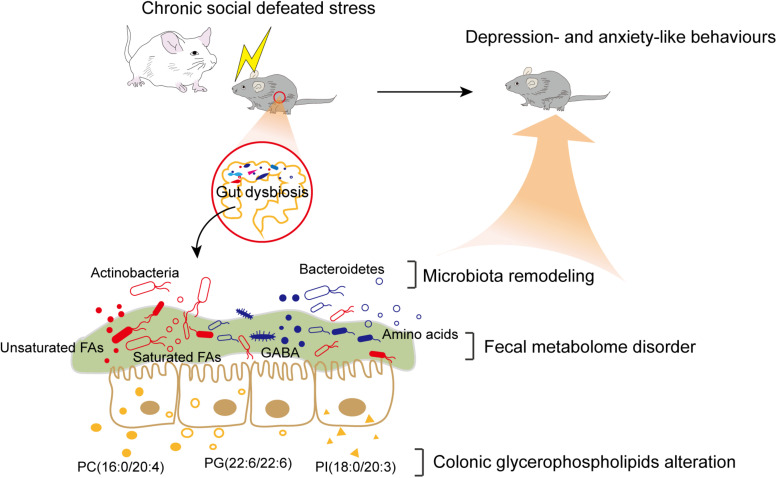
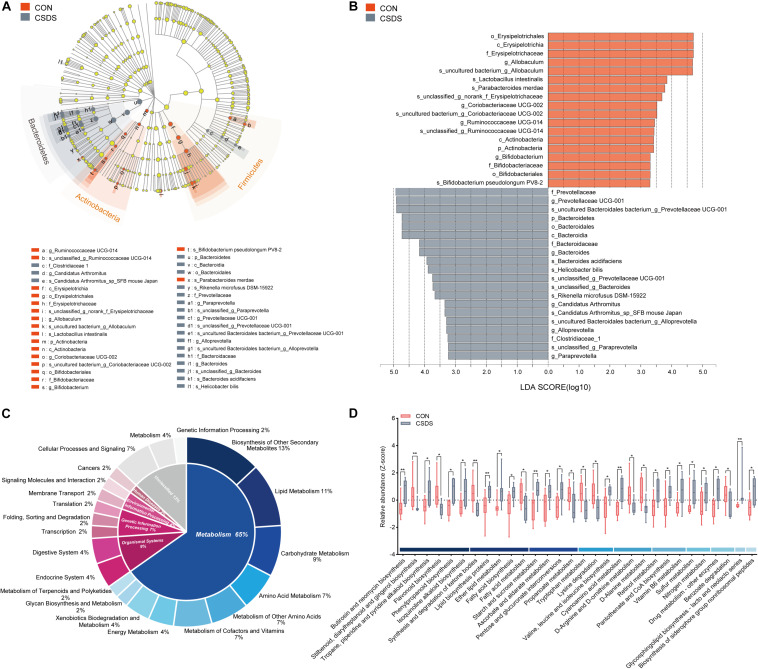
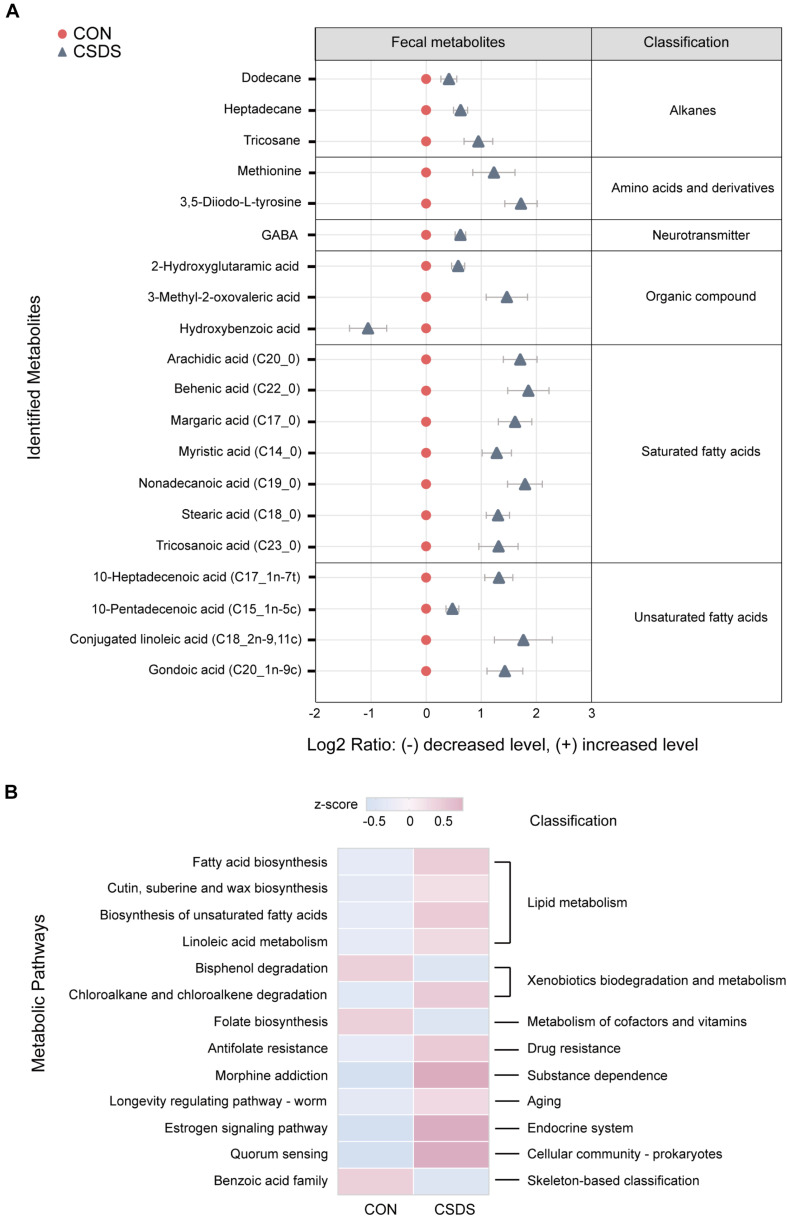
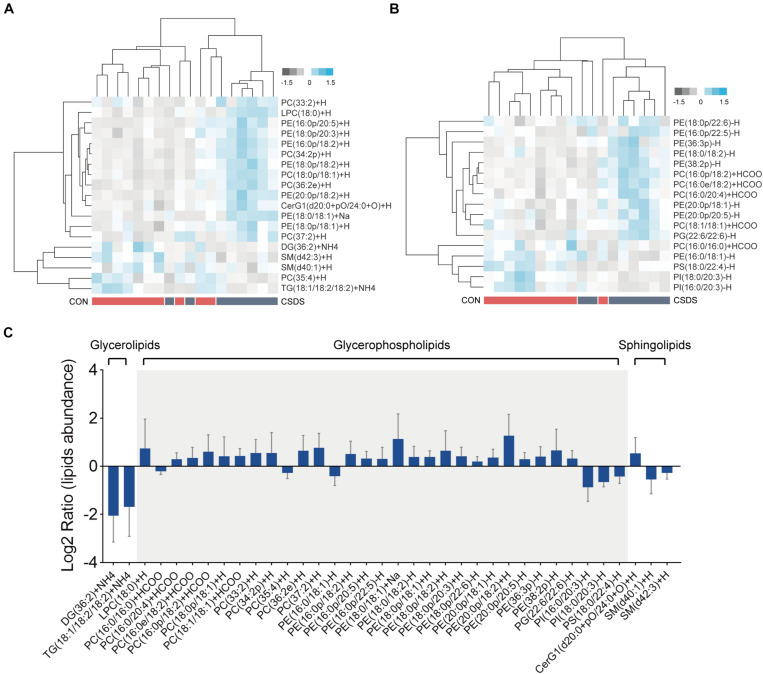
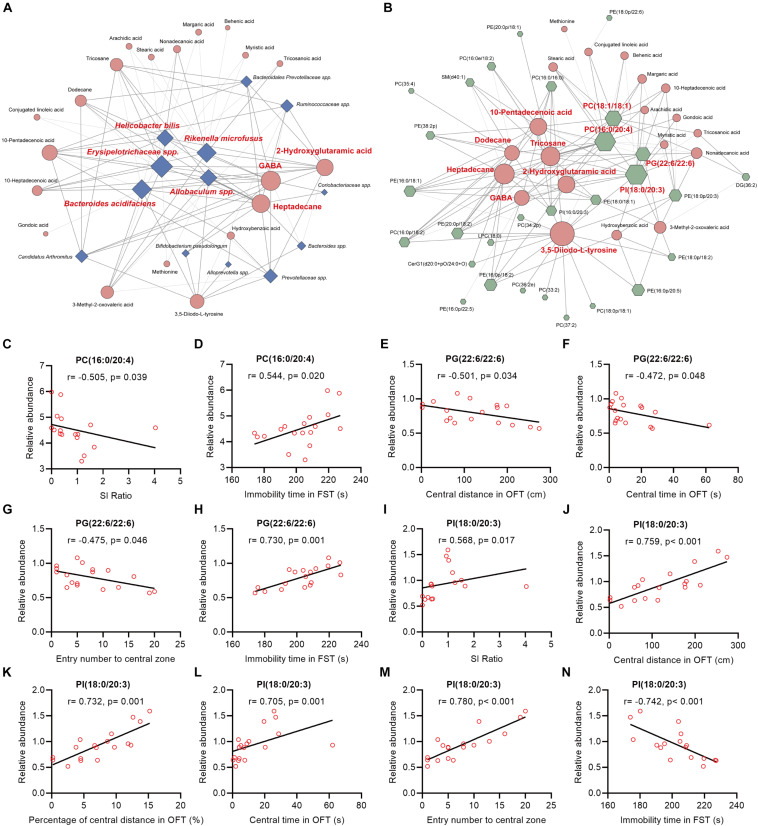
The microbiota-gut-brain axis has been considered to play an important role in the development of depression, but the underlying mechanism remains unclear. The gastrointestinal tract is home to trillions of microbiota and the colon is considered an important site for the interaction between microbiota and host, but few studies have been conducted to evaluate the alterations in the colon. Accordingly, in this study, we established a chronic social defeated stress (CSDS) mice model of depression. We applied 16S rRNA gene sequencing to assess the gut microbial composition and gas and liquid chromatography-mass spectroscopy to identify fecal metabolites and colonic lipids, respectively. Meanwhile, we used Spearman's correlation analysis method to evaluate the associations between the gut microbiota, fecal metabolites, colonic lipids, and behavioral index. In total, there were 20 bacterial taxa and 18 bacterial taxa significantly increased and decreased, respectively, in the CSDS mice. Further, microbial functional prediction demonstrated a disturbance of lipid, carbohydrate, and amino acid metabolism in the CSDS mice. We also found 20 differential fecal metabolites and 36 differential colonic lipids (in the category of glycerolipids, glycerophospholipids, and sphingolipids) in the CSDS mice. Moreover, correlation analysis showed that fecal metabolomic signature was associated with the alterations in the gut microbiota composition and colonic lipidomic profile. Of note, three lipids [PC(16:0/20:4), PG(22:6/22:6), and PI(18:0/20:3), all in the category of glycerophospholipids] were significantly associated with anxiety- and depression-like phenotypes in mice. Taken together, our results indicated that the gut microbiota might be involved in the pathogenesis of depression via influencing fecal metabolites and colonic glycerophospholipid metabolism.
微生物群-肠-脑轴被认为在抑郁症的发生发展中起重要作用,但其潜在机制仍不清楚。胃肠道是数万亿微生物群的家园,结肠被认为是微生物群与宿主相互作用的重要部位,但很少有研究评估结肠中的变化。因此,在本研究中,我们建立了抑郁症的慢性社会挫败应激(CSDS)小鼠模型。我们应用16S rRNA基因测序来评估肠道微生物组成,并用气相色谱-质谱联用和液相色谱-质谱联用分别鉴定粪便代谢物和结肠脂质。同时,我们使用Spearman相关性分析方法来评估肠道微生物群、粪便代谢物、结肠脂质和行为指标之间的关联。总的来说,CSDS小鼠中有20个细菌分类群显著增加,18个细菌分类群显著减少。此外,微生物功能预测表明CSDS小鼠的脂质、碳水化合物和氨基酸代谢受到干扰。我们还在CSDS小鼠中发现了20种差异粪便代谢物和36种差异结肠脂质(甘油olipids、甘油磷脂和鞘脂类)。此外,相关性分析表明粪便代谢组学特征与肠道微生物群组成和结肠脂质组学谱的变化有关。值得注意的是,三种脂质[PC(16:0/20:4)、PG(22:6/22:6)和PI(18:0/20:3),均属于甘油磷脂类]与小鼠的焦虑和抑郁样表型显著相关。综上所述,我们的结果表明肠道微生物群可能通过影响粪便代谢物和结肠甘油磷脂代谢参与抑郁症的发病机制。