Gebreyesus Grum, Lund Mogens Sandø, Sahana Goutam, Su Guosheng
Center for Quantitative Genetics and Genomics, Aarhus University, Tjele, Denmark.
Front Genet. 2021 Jul 19;12:667300. doi: 10.3389/fgene.2021.667300. eCollection 2021.
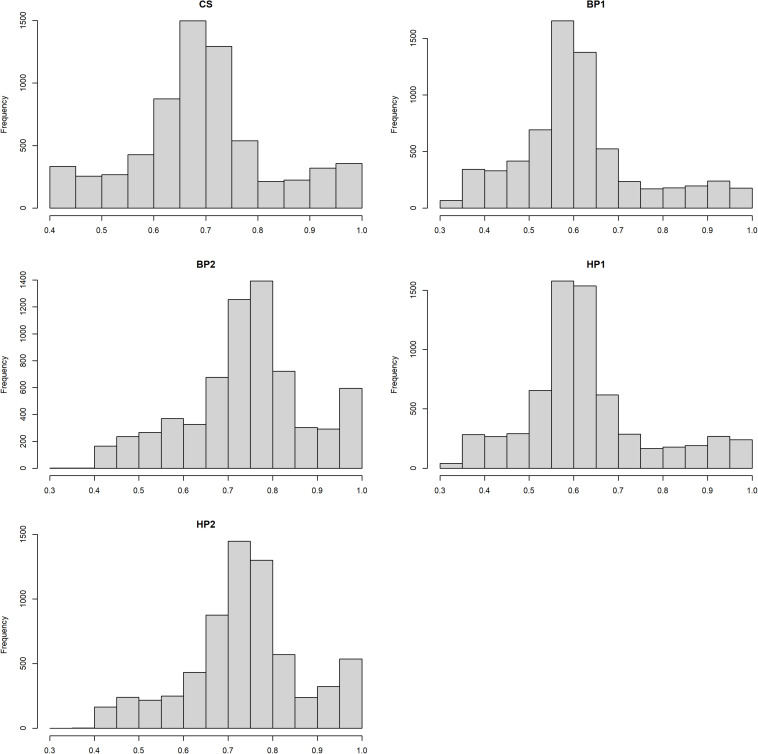
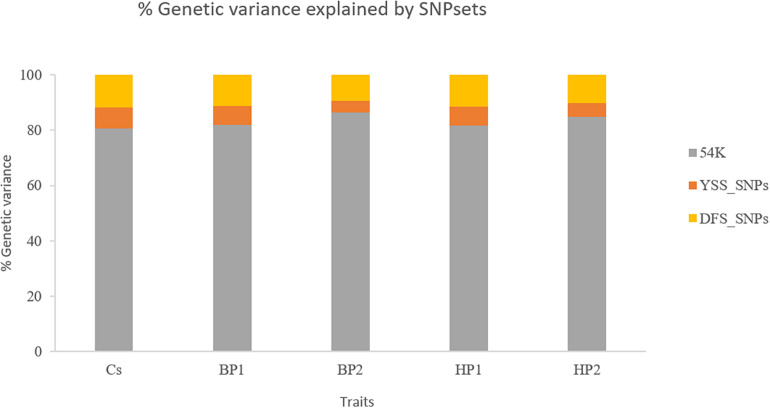
This study investigated effects of integrating single-nucleotide polymorphisms (SNPs) selected based on previous genome-wide association studies (GWASs), from imputed whole-genome sequencing (WGS) data, in the conventional 54K chip on genomic prediction reliability of young stock survival (YSS) traits in dairy cattle. The WGS SNPs included two groups of SNP sets that were selected based on GWAS in the Danish Holstein for YSS index (YSS_SNPs, = 98) and SNPs chosen as peaks of quantitative trait loci for the traits of Nordic total merit index in Denmark-Finland-Sweden dairy cattle populations (DFS_SNPs, = 1,541). Additionally, the study also investigated the possibility of improving genomic prediction reliability for survival traits by modeling the SNPs within recessive lethal haplotypes (LET_SNP, n = 130) detected from the 54K chip in the Nordic Holstein. De-regressed proofs (DRPs) were obtained from 6,558 Danish Holstein bulls genotyped with either 54K chip or customized LD chip that includes SNPs in the standard LD chip and some of the selected WGS SNPs. The chip data were subsequently imputed to 54K SNP together with the selected WGS SNPs. Genomic best linear unbiased prediction (GBLUP) models were implemented to predict breeding values through either pooling the 54K and selected WGS SNPs together as one genetic component (a one-component model) or considering 54K SNPs and selected WGS SNPs as two separate genetic components (a two-component model). Across all the traits, inclusion of each of the selected WGS SNP sets led to negligible improvements in prediction accuracies (0.17 percentage points on average) compared to prediction using only 54K. Similarly, marginal improvement in prediction reliability was obtained when all the selected WGS SNPs were included (0.22 percentage points). No further improvement in prediction reliability was observed when considering random regression on genotype code of recessive lethal alleles in the model including both groups of the WGS SNPs. Additionally, there was no difference in prediction reliability from integrating the selected WGS SNP sets through the two-component model compared to the one-component GBLUP.
本研究调查了将基于既往全基因组关联研究(GWAS)从推算的全基因组测序(WGS)数据中选择的单核苷酸多态性(SNP)整合到传统的54K芯片中,对奶牛幼畜存活(YSS)性状基因组预测可靠性的影响。WGS SNP包括两组SNP集,一组是基于丹麦荷斯坦牛YSS指数的GWAS选择的(YSS_SNP,n = 98),另一组是在丹麦-芬兰-瑞典奶牛群体的北欧总效益指数性状中被选为数量性状基因座峰值的SNP(DFS_SNP,n = 1,541)。此外,该研究还调查了通过对北欧荷斯坦牛54K芯片中检测到的隐性致死单倍型内的SNP进行建模(LET_SNP,n = 130)来提高存活性状基因组预测可靠性的可能性。从6558头用54K芯片或定制LD芯片(包括标准LD芯片中的SNP和一些选定的WGS SNP)进行基因分型的丹麦荷斯坦公牛中获得去回归证明(DRP)。随后将芯片数据与选定的WGS SNP一起推算为54K SNP。实施基因组最佳线性无偏预测(GBLUP)模型,通过将54K和选定的WGS SNP合并为一个遗传成分(单成分模型)或考虑54K SNP和选定的WGS SNP为两个独立的遗传成分(双成分模型)来预测育种值。在所有性状中,与仅使用54K进行预测相比,纳入每个选定的WGS SNP集导致预测准确性的提高可忽略不计(平均0.17个百分点)。同样,当纳入所有选定的WGS SNP时,预测可靠性有小幅提高(0.22个百分点)。在包括两组WGS SNP的模型中,考虑对隐性致死等位基因的基因型编码进行随机回归时,未观察到预测可靠性有进一步提高。此外,与单成分GBLUP相比,通过双成分模型整合选定的WGS SNP集在预测可靠性方面没有差异。