Cooper Isabella D, Brookler Kenneth H, Kyriakidou Yvoni, Elliott Bradley T, Crofts Catherine A P
Translational Physiology Research Group, School of Life Sciences, University of Westminster, 115 New Cavendish Street, London W1W 6UW, UK.
Research Collaborator, Aerospace Medicine and Vestibular Research Laboratory, Mayo Clinic, Scottsdale, AZ 85259, USA.
Biomedicines. 2021 Jul 9;9(7):800. doi: 10.3390/biomedicines9070800.
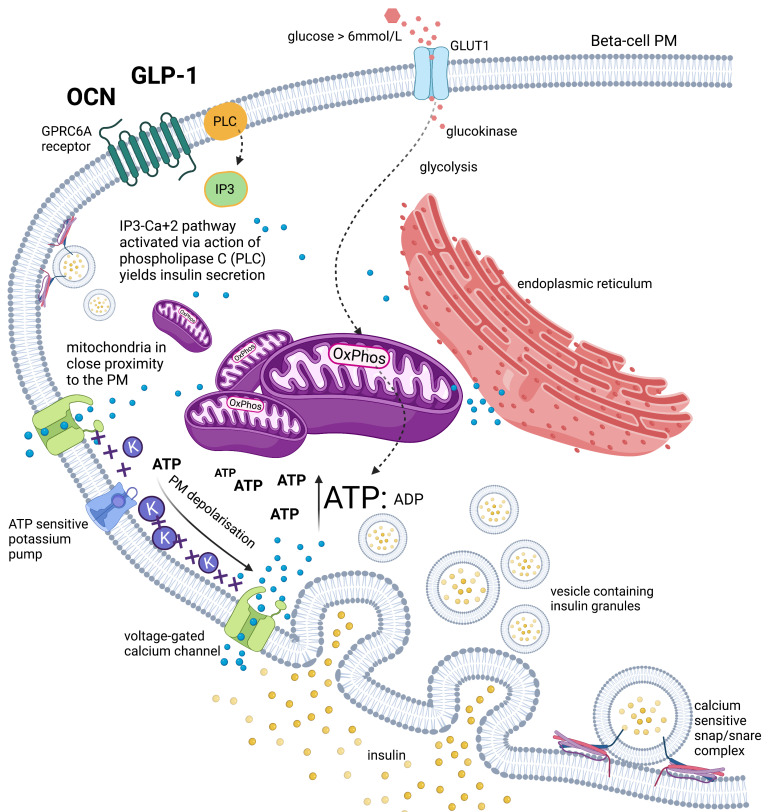
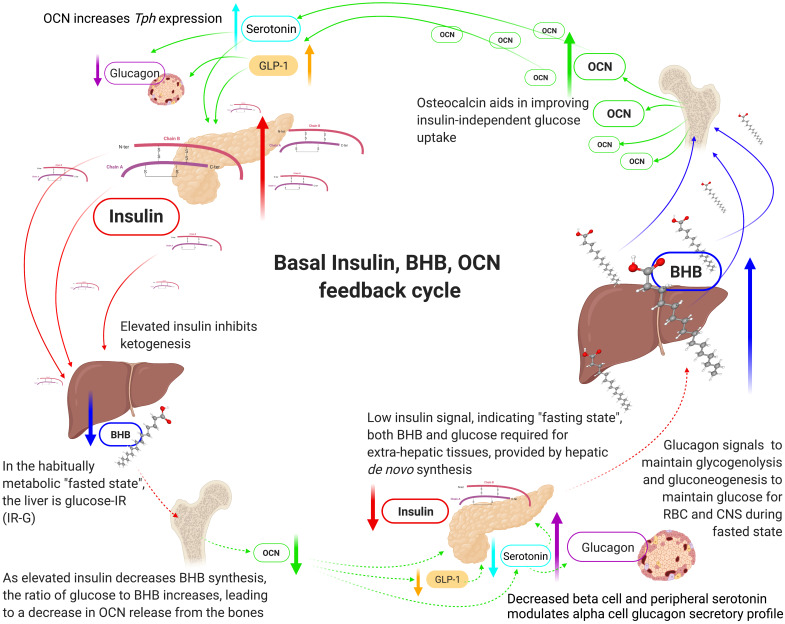
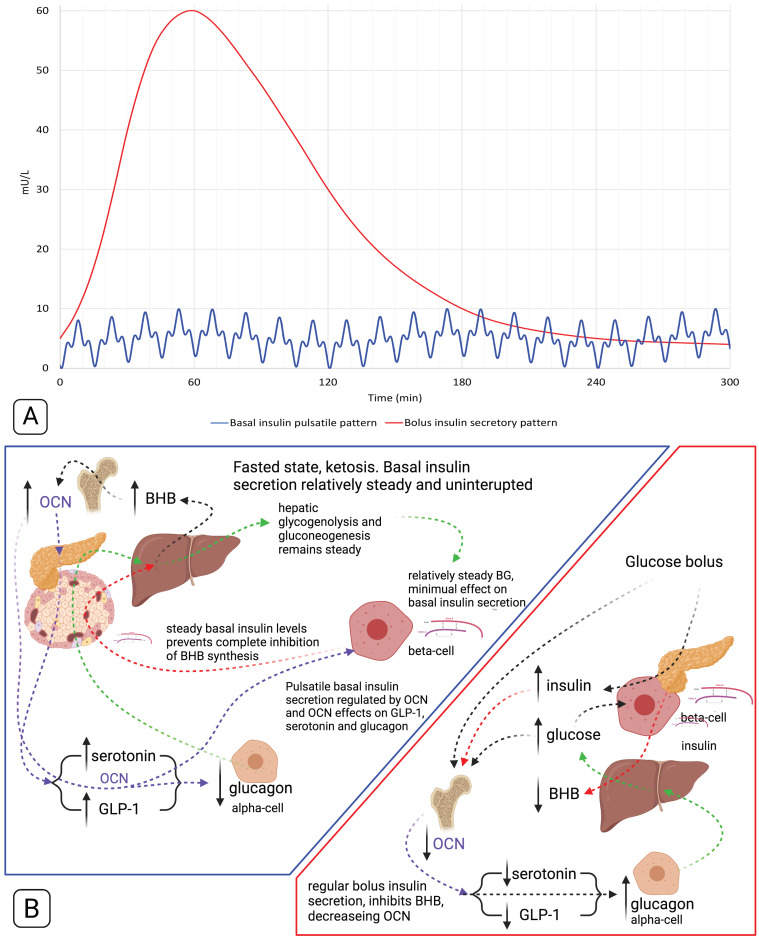
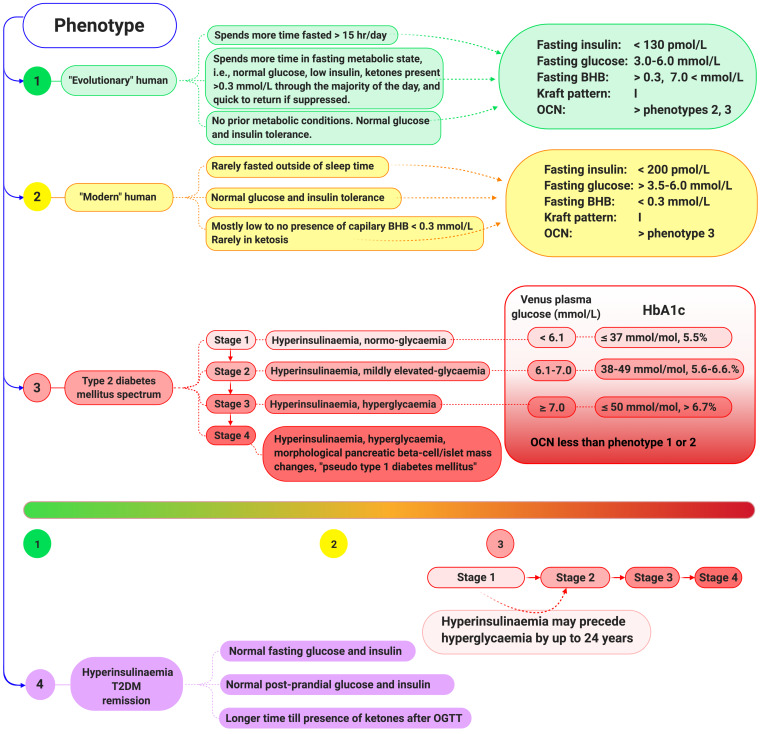
Unlike bolus insulin secretion mechanisms, basal insulin secretion is poorly understood. It is essential to elucidate these mechanisms in non-hyperinsulinaemia healthy persons. This establishes a baseline for investigation into pathologies where these processes are dysregulated, such as in type 2 diabetes (T2DM), cardiovascular disease (CVD), certain cancers and dementias. Chronic hyperinsulinaemia enforces glucose fueling, depleting the NAD+ dependent antioxidant activity that increases mitochondrial reactive oxygen species (mtROS). Consequently, beta-cell mitochondria increase uncoupling protein expression, which decreases the mitochondrial ATP surge generation capacity, impairing bolus mediated insulin exocytosis. Excessive ROS increases the Drp1:Mfn2 ratio, increasing mitochondrial fission, which increases mtROS; endoplasmic reticulum-stress and impaired calcium homeostasis ensues. Healthy individuals in habitual ketosis have significantly lower glucagon and insulin levels than T2DM individuals. As beta-hydroxybutyrate rises, hepatic gluconeogenesis and glycogenolysis supply extra-hepatic glucose needs, and osteocalcin synthesis/release increases. We propose insulin's primary role is regulating beta-hydroxybutyrate synthesis, while the role of bone regulates glucose uptake sensitivity via osteocalcin. Osteocalcin regulates the alpha-cell glucagon secretory profile via glucagon-like peptide-1 and serotonin, and beta-hydroxybutyrate synthesis via regulating basal insulin levels. Establishing metabolic phenotypes aids in resolving basal insulin secretion regulation, enabling elucidation of the pathological changes that occur and progress into chronic diseases associated with ageing.
与胰岛素脉冲式分泌机制不同,基础胰岛素分泌的相关机制目前仍知之甚少。阐明非高胰岛素血症健康人群的这些机制至关重要。这为研究这些过程失调的病理学情况建立了一个基线,比如2型糖尿病(T2DM)、心血管疾病(CVD)、某些癌症和痴呆症。慢性高胰岛素血症会促使葡萄糖供能,消耗依赖烟酰胺腺嘌呤二核苷酸(NAD+)的抗氧化活性,从而增加线粒体活性氧(mtROS)。因此,β细胞线粒体会增加解偶联蛋白的表达,这会降低线粒体ATP激增的生成能力,损害脉冲式介导的胰岛素胞吐作用。过量的活性氧会增加动力相关蛋白1(Drp1)与线粒体融合蛋白2(Mfn2)的比例,增加线粒体裂变,进而增加线粒体活性氧;随之会出现内质网应激和钙稳态受损。习惯性处于酮症状态的健康个体的胰高血糖素和胰岛素水平明显低于2型糖尿病患者。随着β-羟基丁酸水平升高,肝脏糖异生和糖原分解可满足肝外葡萄糖需求,骨钙素的合成/释放也会增加。我们认为胰岛素的主要作用是调节β-羟基丁酸的合成,而骨骼则通过骨钙素调节葡萄糖摄取敏感性。骨钙素通过胰高血糖素样肽-1和血清素调节α细胞的胰高血糖素分泌模式,并通过调节基础胰岛素水平来调节β-羟基丁酸的合成。确定代谢表型有助于解析基础胰岛素分泌调节机制,从而阐明所发生的病理变化以及与衰老相关的慢性疾病的发展进程。