Biodesign Center for Environmental Health Engineering, Biodesign Institute, Arizona State University, Tempe, AZ, 85287, USA.
Biodesign Center for Personalized Diagnostics, Biodesign Institute, Arizona State University, Tempe, AZ, 85287, USA.
BMC Infect Dis. 2021 Aug 13;21(1):810. doi: 10.1186/s12879-021-06526-5.
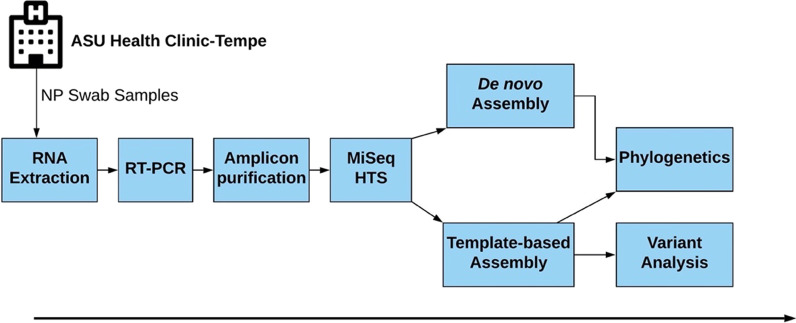
Local transmission of seasonal influenza viruses (IVs) can be difficult to resolve. Here, we study if coupling high-throughput sequencing (HTS) of hemagglutinin (HA) and neuraminidase (NA) genes with variant analysis can resolve strains from local transmission that have identical consensus genome. We analyzed 24 samples collected over four days in January 2020 at a large university in the US. We amplified complete hemagglutinin (HA) and neuraminidase (NA) genomic segments followed by Illumina sequencing. We identified consensus complete HA and NA segments using BLASTn and performed variant analysis on strains whose HA and NA segments were 100% similar.
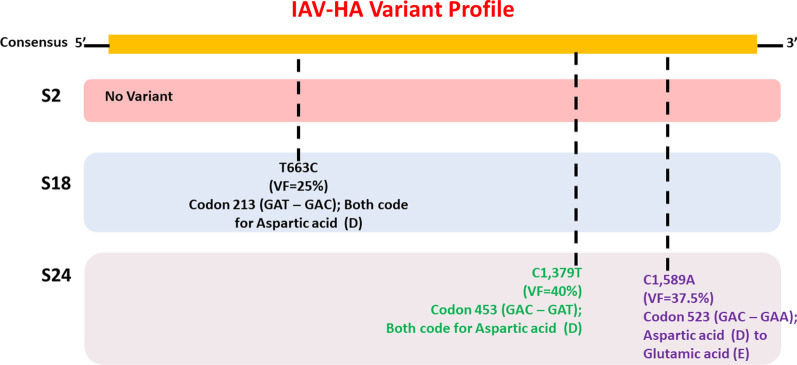
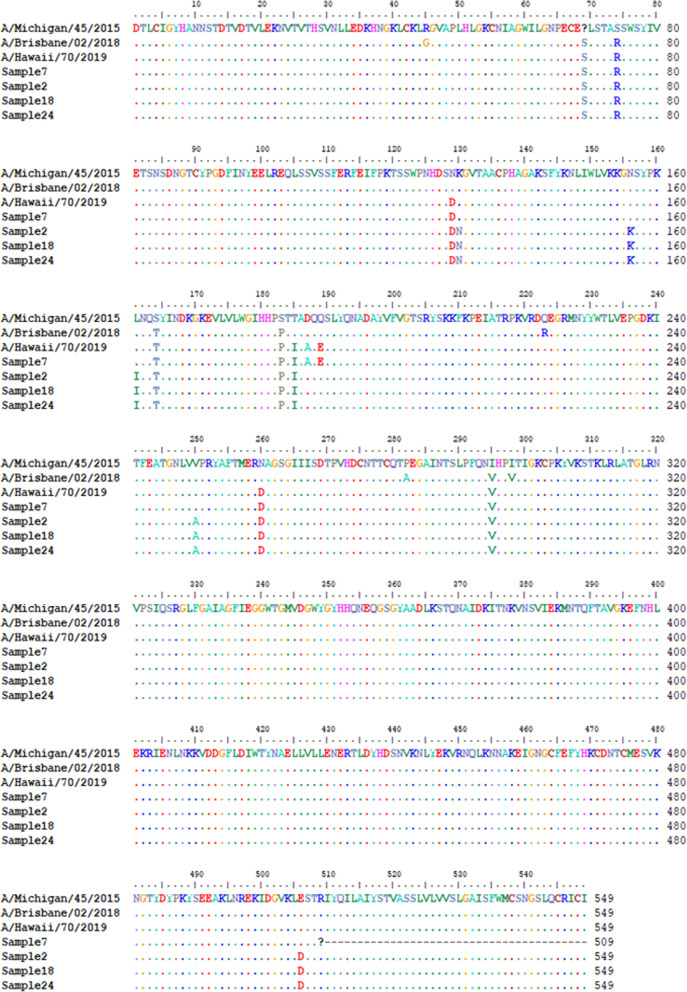
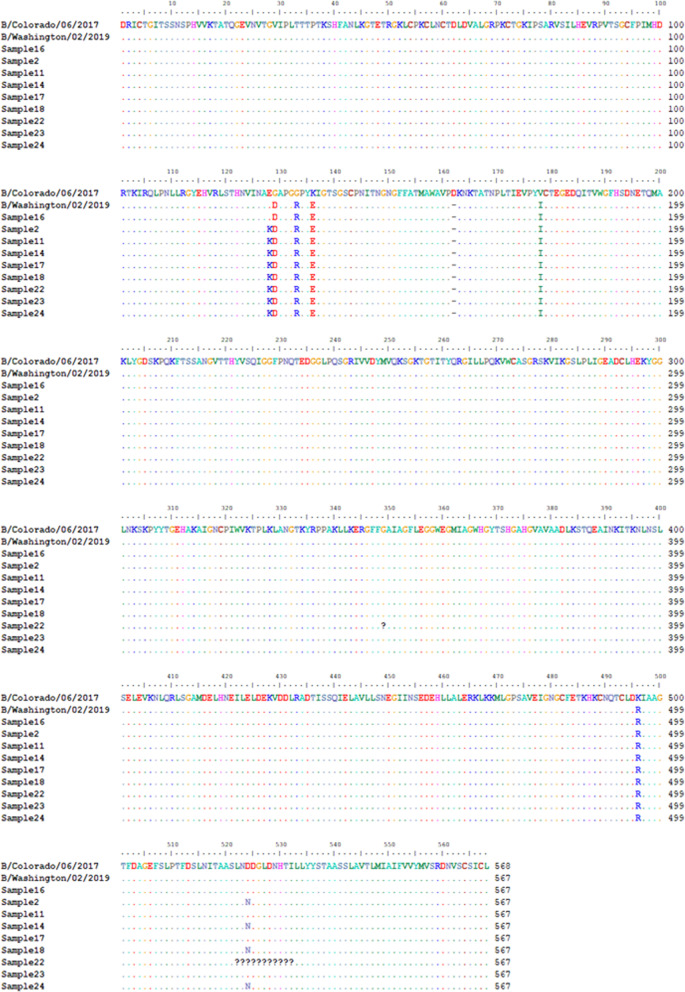
Twelve of the 24 samples were PCR positive, and we detected complete HA and/or NA segments by de novo assembly in 83.33% (10/12) of them. Similarity and phylogenetic analysis showed that 70% (7/10) of the strains were distinct while the remaining 30% had identical consensus sequences. These three samples also had IAV and IBV co-infection. However, subsequent variant analysis showed that they had distinct variant profiles. While the IAV HA of one sample had no variant, another had a T663C mutation and another had both C1379T and C1589A.
In this study, we showed that HTS coupled with variant analysis of only HA and NA genes can help resolve variants that are closely related. We also provide evidence that during a short time period in the 2019-2020 season, co-infection of IAV and IBV occurred on the university campus and both 2020/2021 and 2021/2022 WHO recommended H1N1 vaccine strains were co-circulating.
季节性流感病毒(IVs)的本地传播可能难以解决。在这里,我们研究是否可以将血凝素(HA)和神经氨酸酶(NA)基因的高通量测序(HTS)与变异分析相结合,解决具有相同共识基因组的本地传播菌株。我们分析了 2020 年 1 月在美国一所大型大学四天内采集的 24 个样本。我们扩增了完整的血凝素(HA)和神经氨酸酶(NA)基因组片段,然后进行 Illumina 测序。我们使用 BLASTn 确定了共识完整 HA 和 NA 片段,并对 HA 和 NA 片段完全相似的菌株进行了变异分析。
24 个样本中有 12 个 PCR 阳性,我们在其中 83.33%(10/12)的样本中通过从头组装检测到完整的 HA 和/或 NA 片段。相似性和系统发育分析表明,70%(7/10)的菌株是不同的,而其余 30%的菌株具有相同的共识序列。这三个样本还同时感染了甲型流感病毒和乙型流感病毒。然而,随后的变异分析表明它们具有不同的变异特征。一个样本的甲型流感病毒 HA 没有变异,另一个样本有 T663C 突变,另一个样本则同时有 C1379T 和 C1589A 突变。
在这项研究中,我们表明仅通过 HA 和 NA 基因的 HTS 结合变异分析可以帮助解决密切相关的变异。我们还提供了证据,即在 2019-2020 季节的短时间内,甲型流感病毒和乙型流感病毒同时在大学校园内感染,并且 2020/2021 和 2021/2022 年世界卫生组织推荐的 H1N1 疫苗株同时循环。