Mervin Lewis H, Trapotsi Maria-Anna, Afzal Avid M, Barrett Ian P, Bender Andreas, Engkvist Ola
Molecular AI, Discovery Sciences, R&D, AstraZeneca, Cambridge, UK.
Department of Chemistry, Centre for Molecular Informatics, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK.
J Cheminform. 2021 Aug 19;13(1):62. doi: 10.1186/s13321-021-00539-7.
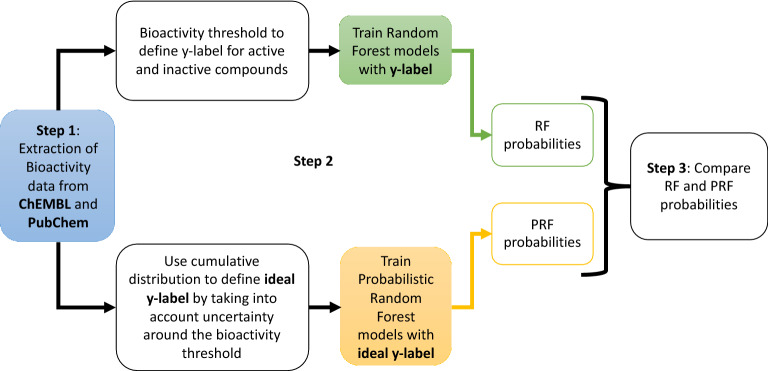
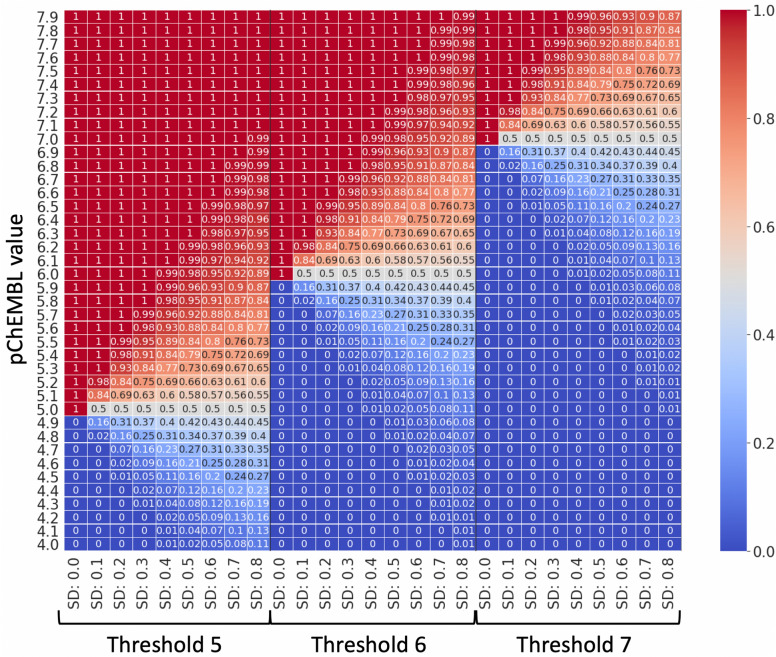
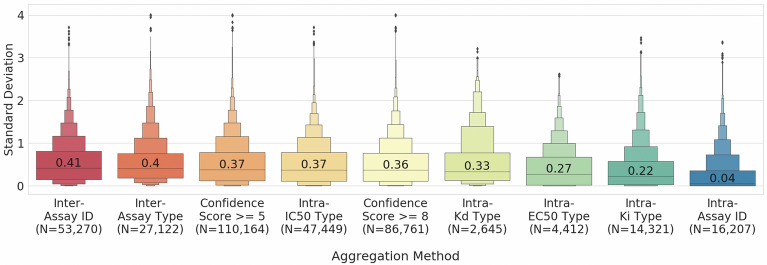
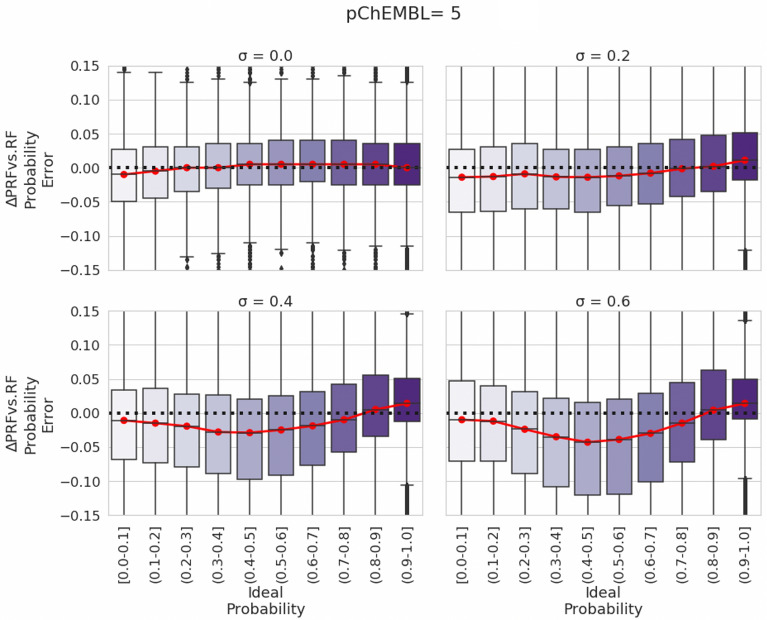
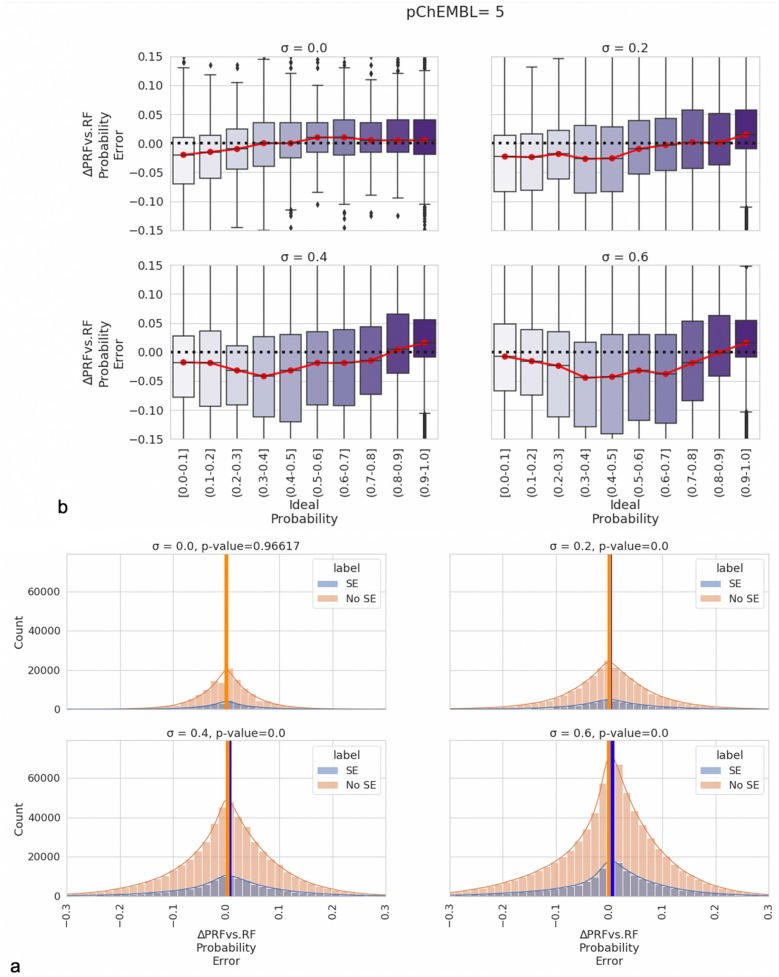
Measurements of protein-ligand interactions have reproducibility limits due to experimental errors. Any model based on such assays will consequentially have such unavoidable errors influencing their performance which should ideally be factored into modelling and output predictions, such as the actual standard deviation of experimental measurements (σ) or the associated comparability of activity values between the aggregated heterogenous activity units (i.e., K versus IC values) during dataset assimilation. However, experimental errors are usually a neglected aspect of model generation. In order to improve upon the current state-of-the-art, we herein present a novel approach toward predicting protein-ligand interactions using a Probabilistic Random Forest (PRF) classifier. The PRF algorithm was applied toward in silico protein target prediction across ~ 550 tasks from ChEMBL and PubChem. Predictions were evaluated by taking into account various scenarios of experimental standard deviations in both training and test sets and performance was assessed using fivefold stratified shuffled splits for validation. The largest benefit in incorporating the experimental deviation in PRF was observed for data points close to the binary threshold boundary, when such information was not considered in any way in the original RF algorithm. For example, in cases when σ ranged between 0.4-0.6 log units and when ideal probability estimates between 0.4-0.6, the PRF outperformed RF with a median absolute error margin of ~ 17%. In comparison, the baseline RF outperformed PRF for cases with high confidence to belong to the active class (far from the binary decision threshold), although the RF models gave errors smaller than the experimental uncertainty, which could indicate that they were overtrained and/or over-confident. Finally, the PRF models trained with putative inactives decreased the performance compared to PRF models without putative inactives and this could be because putative inactives were not assigned an experimental pXC value, and therefore they were considered inactives with a low uncertainty (which in practice might not be true). In conclusion, PRF can be useful for target prediction models in particular for data where class boundaries overlap with the measurement uncertainty, and where a substantial part of the training data is located close to the classification threshold.
由于实验误差,蛋白质 - 配体相互作用的测量存在可重复性限制。基于此类测定的任何模型都会不可避免地存在影响其性能的误差,理论上在建模和输出预测中应考虑这些误差,例如实验测量的实际标准偏差(σ)或在数据集同化期间聚集的异质活性单位之间活性值的相关可比性(即K值与IC值)。然而,实验误差通常是模型生成中被忽视的一个方面。为了改进当前的技术水平,我们在此提出一种使用概率随机森林(PRF)分类器预测蛋白质 - 配体相互作用的新方法。PRF算法被应用于跨ChEMBL和PubChem中约550个任务的计算机模拟蛋白质靶点预测。通过考虑训练集和测试集中实验标准偏差的各种情况来评估预测,并使用五重分层随机拆分进行验证来评估性能。当原始随机森林(RF)算法完全不考虑此类信息时,在PRF中纳入实验偏差对接近二元阈值边界的数据点观察到最大益处。例如,当σ在0.4 - 0.6对数单位范围内且理想概率估计在0.4 - 0.6之间时,PRF的表现优于RF,中位数绝对误差幅度约为17%。相比之下,对于高度确信属于活性类(远离二元决策阈值)的情况,基线RF的表现优于PRF,尽管RF模型给出的误差小于实验不确定性,这可能表明它们过度训练和/或过度自信。最后,与没有假定非活性物质的PRF模型相比,用假定非活性物质训练的PRF模型性能下降,这可能是因为假定非活性物质没有被赋予实验pXC值,因此它们被视为不确定性低的非活性物质(而在实际中可能并非如此)。总之,PRF对于靶点预测模型可能是有用的,特别是对于类别边界与测量不确定性重叠且大部分训练数据位于分类阈值附近的数据。