Institute for Disease Modeling, Bill & Melinda Gates Foundation, Seattle, Washington, USA.
Centre for Health Informatics, Computing, and Statistics, Lancaster University, Lancaster, United Kingdom.
Clin Infect Dis. 2022 Jun 10;74(11):1993-2000. doi: 10.1093/cid/ciab745.
Diverse environmental exposures and risk factors have been implicated in the transmission of Salmonella Typhi, but the dominant transmission pathways through the environment to susceptible humans remain unknown. Here, we use spatial, bacterial genomic, and hydrological data to refine our view of typhoid transmission in an endemic setting.
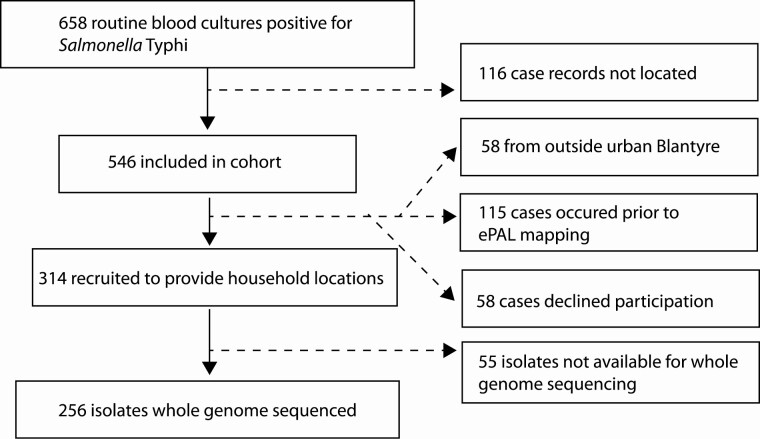
A total of 546 patients presenting to Queen Elizabeth Central Hospital in Blantyre, Malawi, with blood culture-confirmed typhoid fever between April 2015 and January 2017 were recruited to a cohort study. The households of a subset of these patients were geolocated, and 256 S. Typhi isolates were whole-genome sequenced. Pairwise single-nucleotide variant distances were incorporated into a geostatistical modeling framework using multidimensional scaling.
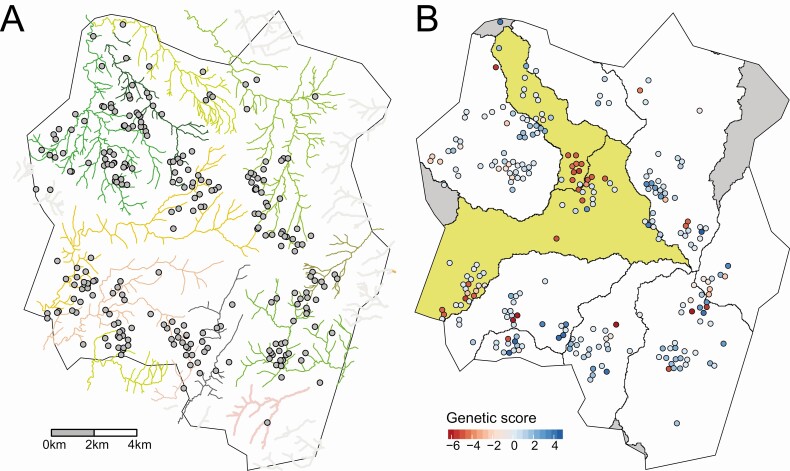
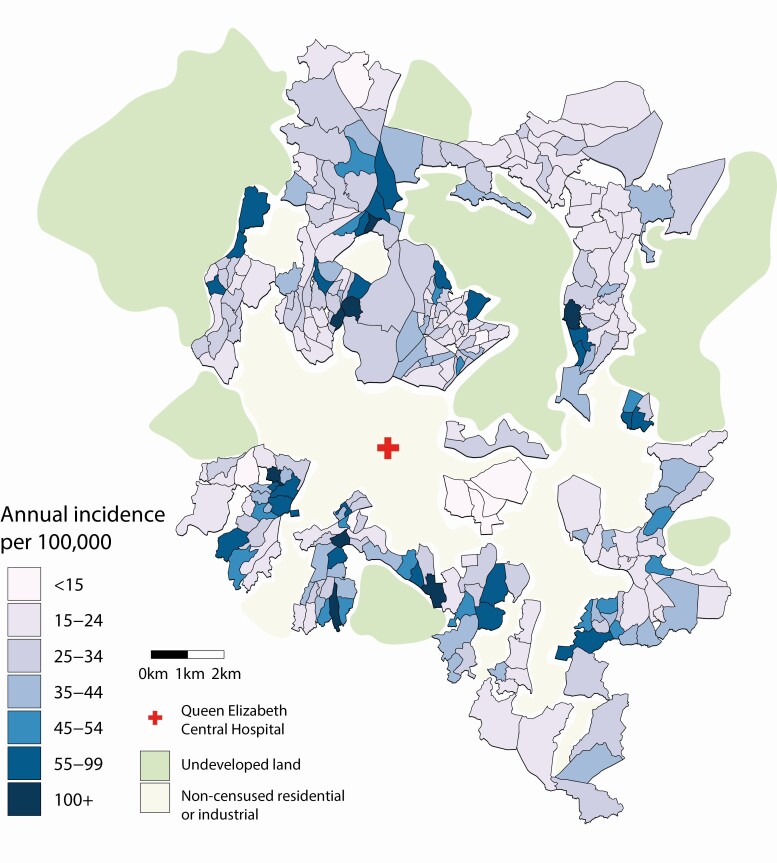
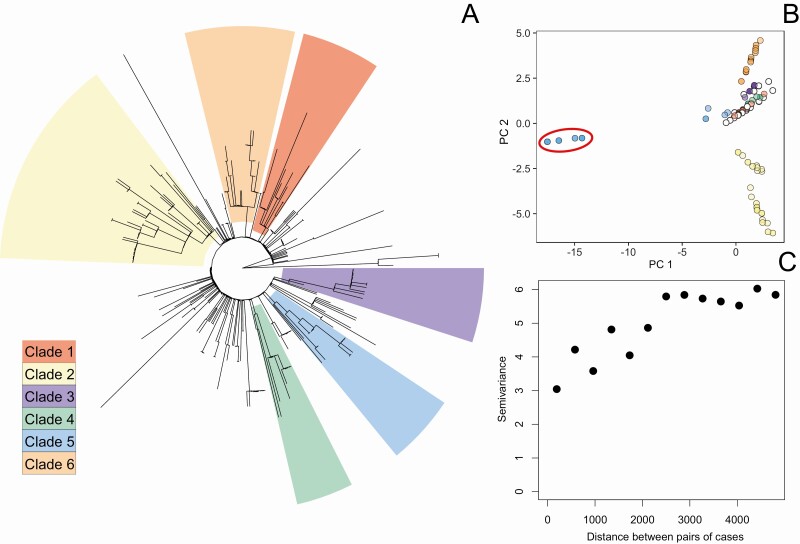
Typhoid fever was not evenly distributed across Blantyre, with estimated minimum incidence ranging across the city from <15 to >100 cases per 100 000 population per year. Pairwise single-nucleotide variant distance and physical household distances were significantly correlated (P = .001). We evaluated the ability of river catchment to explain the spatial patterns of genomics observed, finding that it significantly improved the fit of the model (P = .003). We also found spatial correlation at a smaller spatial scale, of households living <192 m apart.
These findings reinforce the emerging view that hydrological systems play a key role in the transmission of typhoid fever. By combining genomic and spatial data, we show how multifaceted data can be used to identify high incidence areas, explain the connections between them, and inform targeted environmental surveillance, all of which will be critical to shape local and regional typhoid control strategies.
多种环境暴露和风险因素与伤寒沙门氏菌的传播有关,但通过环境传播给易感人群的主要途径仍不清楚。在这里,我们利用空间、细菌基因组和水文学数据来完善我们对地方性环境中伤寒传播的看法。
2015 年 4 月至 2017 年 1 月期间,在马拉维布兰太尔的伊丽莎白女王中央医院就诊的 546 名血培养确诊为伤寒的患者被招募到一个队列研究中。这些患者的一部分家庭进行了地理定位,256 株伤寒沙门氏菌分离株进行了全基因组测序。使用多维尺度分析将成对的单核苷酸变异距离纳入地质统计学建模框架。
伤寒在布兰太尔的分布并不均匀,估计全市每年每 10 万人中有<15 至>100 例病例。成对的单核苷酸变异距离和物理家庭距离呈显著相关(P=0.001)。我们评估了河流集水区解释观察到的基因组学空间模式的能力,发现它显著提高了模型的拟合度(P=0.003)。我们还发现了较小空间尺度上的空间相关性,即居住在相距<192 米的家庭之间存在相关性。
这些发现强化了水文学系统在伤寒传播中起关键作用的观点。通过结合基因组和空间数据,我们展示了多方面的数据如何用于识别高发病率地区,解释它们之间的联系,并为有针对性的环境监测提供信息,所有这些都将对塑造当地和区域的伤寒控制策略至关重要。