Agarwal Gaurav, Choudhary Divya, Stice Shaun P, Myers Brendon K, Gitaitis Ronald D, Venter Stephanus N, Kvitko Brian H, Dutta Bhabesh
Department of Plant Pathology, Coastal Plain Experimental Station, University of Georgia, Tifton, GA, United States.
Department of Plant Pathology, University of Georgia, Athens, GA, United States.
Front Microbiol. 2021 Aug 19;12:684756. doi: 10.3389/fmicb.2021.684756. eCollection 2021.
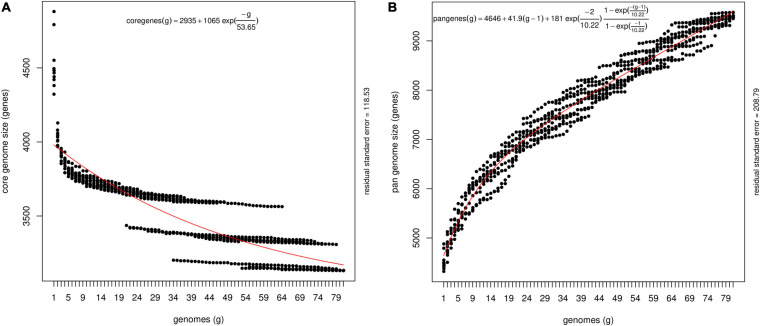
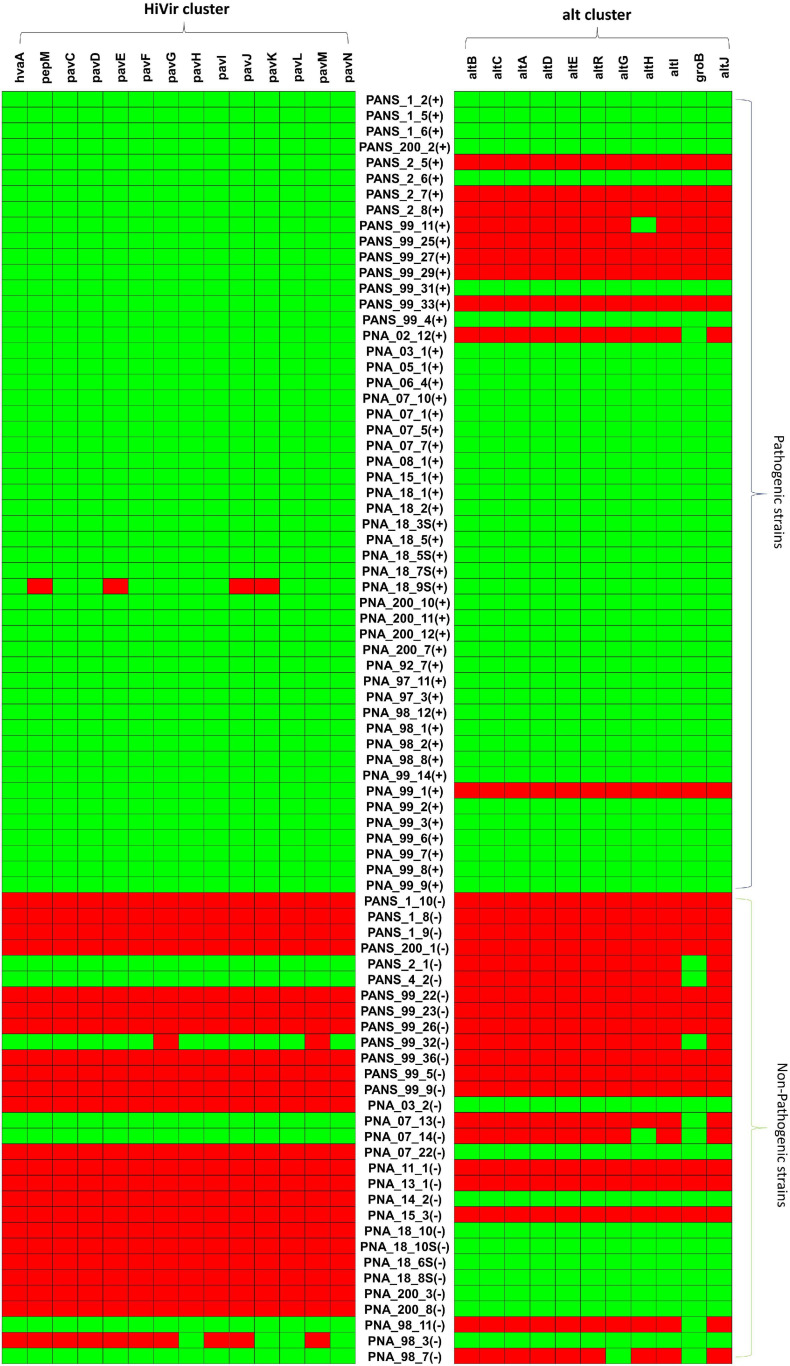
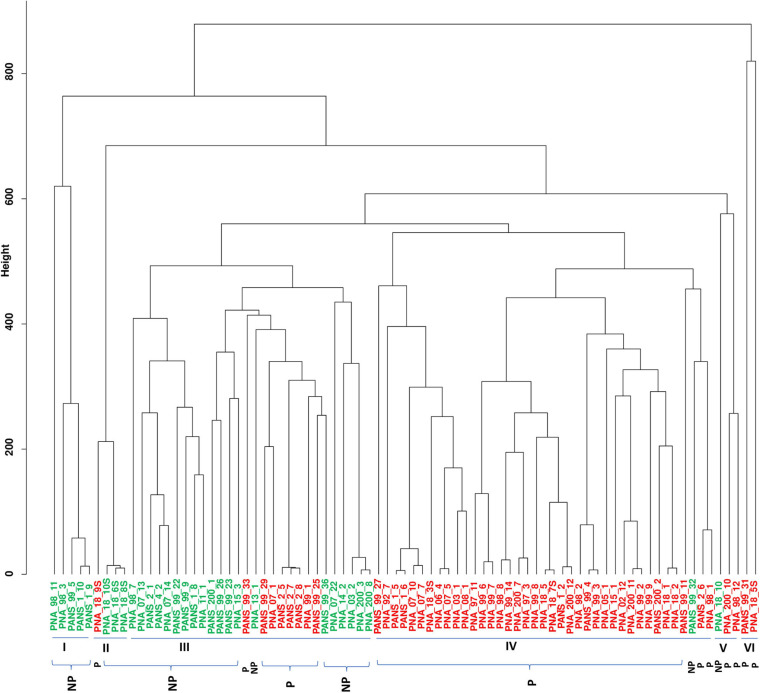
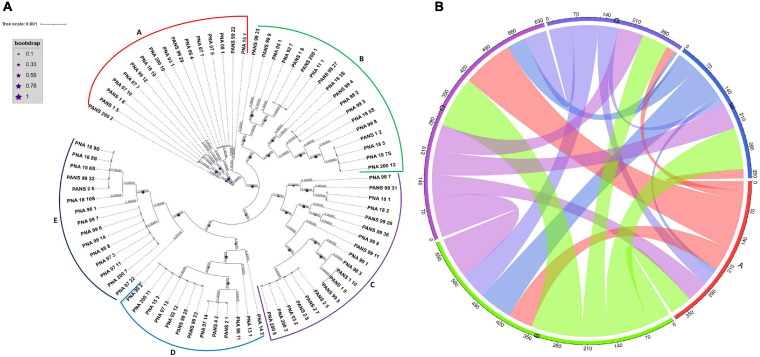
Pantoea ananatis, a gram negative and facultative anaerobic bacterium is a member of a spp. complex that causes center rot of onion, which significantly affects onion yield and quality. This pathogen does not have typical virulence factors like type II or type III secretion systems but appears to require a biosynthetic gene-cluster, HiVir/PASVIL (located chromosomally comprised of 14 genes), for a phosphonate secondary metabolite, and the '' gene cluster (located in plasmid and comprised of 11 genes) that aids in bacterial colonization in onion bulbs by imparting tolerance to thiosulfinates. We conducted a deep pan-genome-wide association study (pan-GWAS) to predict additional genes associated with pathogenicity in using a panel of diverse strains ( = 81). We utilized a red-onion scale necrosis assay as an indicator of pathogenicity. Based on this assay, we differentiated pathogenic ( = 51)- vs. non-pathogenic ( = 30)-strains phenotypically. Pan-genome analysis revealed a large core genome of 3,153 genes and a flexible accessory genome. Pan-GWAS using the presence and absence variants (PAVs) predicted 42 genes, including 14 from the previously identified HiVir/PASVIL cluster associated with pathogenicity, and 28 novel genes that were not previously associated with pathogenicity in onion. Of the 28 novel genes identified, eight have annotated functions of site-specific tyrosine kinase, N-acetylmuramoyl-L-alanine amidase, conjugal transfer, and HTH-type transcriptional regulator. The remaining 20 genes are currently hypothetical. Further, a core-genome SNPs-based phylogeny and horizontal gene transfer (HGT) studies were also conducted to assess the extent of lateral gene transfer among diverse strains. Phylogenetic analysis based on PAVs and whole genome multi locus sequence typing (wgMLST) rather than core-genome SNPs distinguished red-scale necrosis inducing (pathogenic) strains from non-scale necrosis inducing (non-pathogenic) strains of . A total of 1182 HGT events including the HiVir/PASVIL and cluster genes were identified. These events could be regarded as a major contributing factor to the diversification, niche-adaptation and potential acquisition of pathogenicity/virulence genes in .
菠萝泛菌,一种革兰氏阴性兼性厌氧菌,是引起洋葱中心腐烂的一个菌种复合体的成员,这会显著影响洋葱的产量和品质。这种病原体没有典型的毒力因子,如II型或III型分泌系统,但似乎需要一个生物合成基因簇HiVir/PASVIL(位于染色体上,由14个基因组成)来合成一种膦酸盐次级代谢产物,以及“”基因簇(位于质粒上,由11个基因组成),该基因簇通过赋予对硫代亚磺酸盐的耐受性来帮助细菌在洋葱鳞茎中定殖。我们进行了一项全基因组范围的深度关联研究(泛基因组全关联研究,pan-GWAS),以使用一组不同的菌株(n = 81)预测与菠萝泛菌致病性相关的其他基因。我们使用红洋葱鳞片坏死试验作为致病性的指标。基于该试验,我们从表型上区分了致病性菌株(n = 51)和非致病性菌株(n = 30)。泛基因组分析揭示了一个由3153个基因组成的大核心基因组和一个灵活的辅助基因组。使用存在和缺失变异(PAV)进行的泛基因组全关联研究预测了42个基因,其中包括14个来自先前鉴定的与致病性相关的HiVir/PASVIL基因簇,以及28个以前未与洋葱致病性相关的新基因。在鉴定出的28个新基因中,8个具有位点特异性酪氨酸激酶、N-乙酰胞壁酰-L-丙氨酸酰胺酶、接合转移和HTH型转录调节因子的注释功能。其余20个基因目前是假设性的。此外,还进行了基于核心基因组单核苷酸多态性的系统发育和水平基因转移(HGT)研究,以评估不同菠萝泛菌菌株之间横向基因转移的程度。基于PAV和全基因组多位点序列分型(wgMLST)而非核心基因组单核苷酸多态性的系统发育分析区分了引起红鳞片坏死的(致病性)菌株和不引起鳞片坏死的(非致病性)菠萝泛菌菌株。共鉴定出1182个水平基因转移事件,包括HiVir/PASVIL和基因簇基因。这些事件可被视为菠萝泛菌多样化、生态位适应以及致病性/毒力基因潜在获得的主要促成因素。