Farooq Tahir, Umar Muhammad, She Xiaoman, Tang Yafei, He Zifu
Plant Protection Research Institute and Guangdong Provincial Key Laboratory of High Technology for Plant Protection, Guangdong Academy of Agricultural Sciences, Guangzhou 510640, P.R. China.
Tasmanian Institute of Agriculture, New Town Research Laboratories, University of Tasmania, 13 St. Johns Avenue, New Town, TAS 7008, Australia.
Virus Evol. 2021 Jun 4;7(2):veab054. doi: 10.1093/ve/veab054. eCollection 2021.
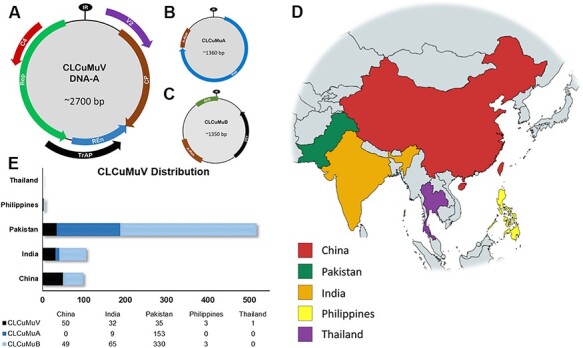
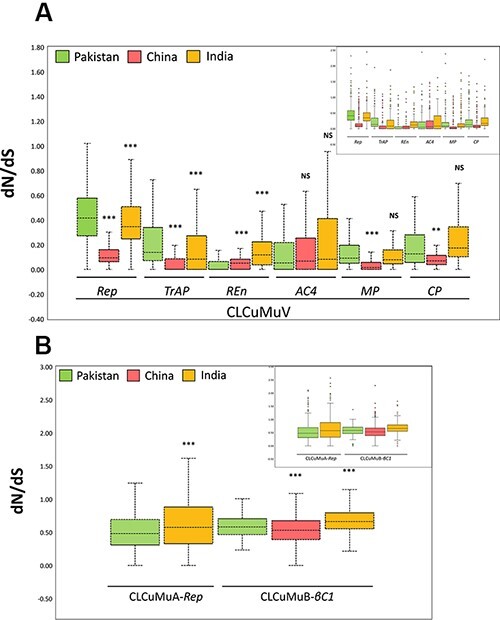
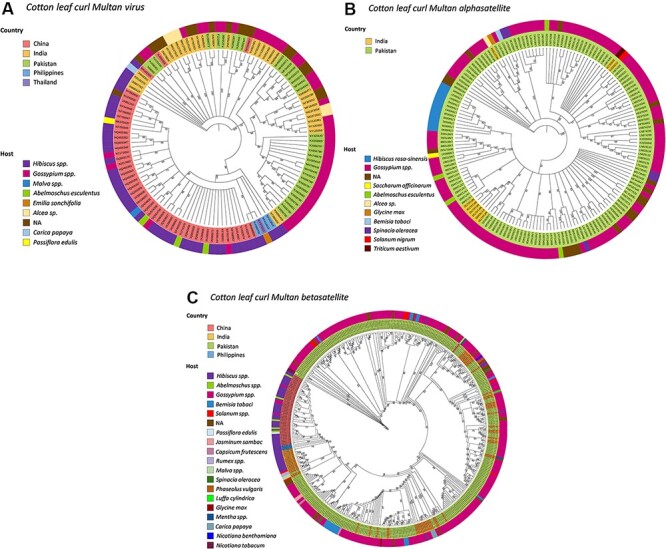
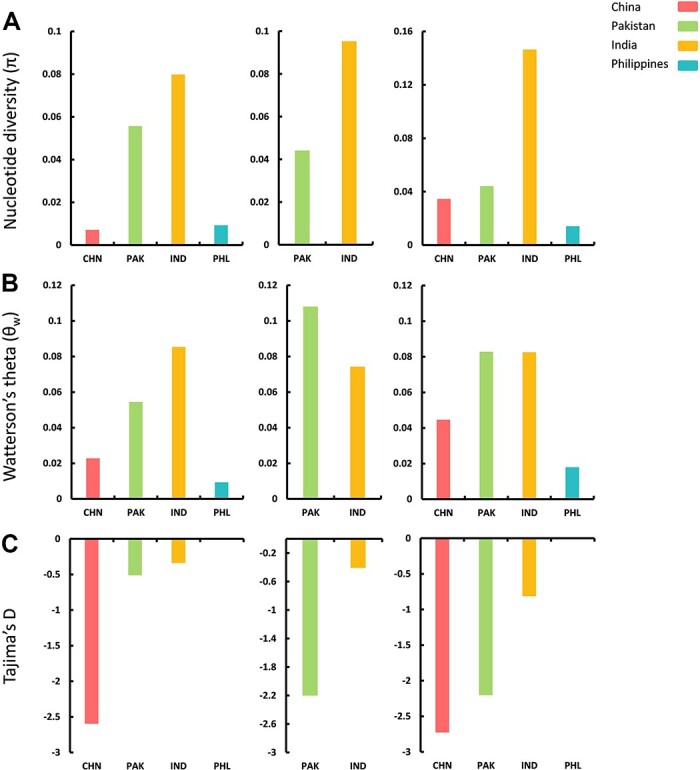
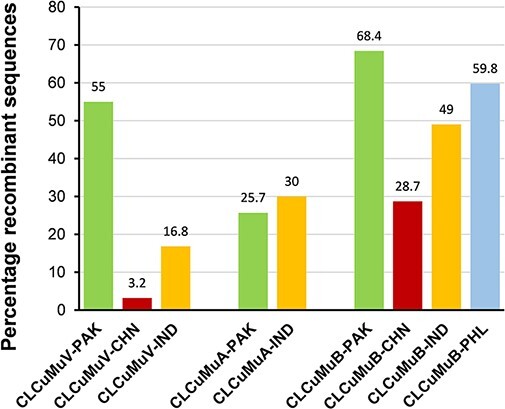
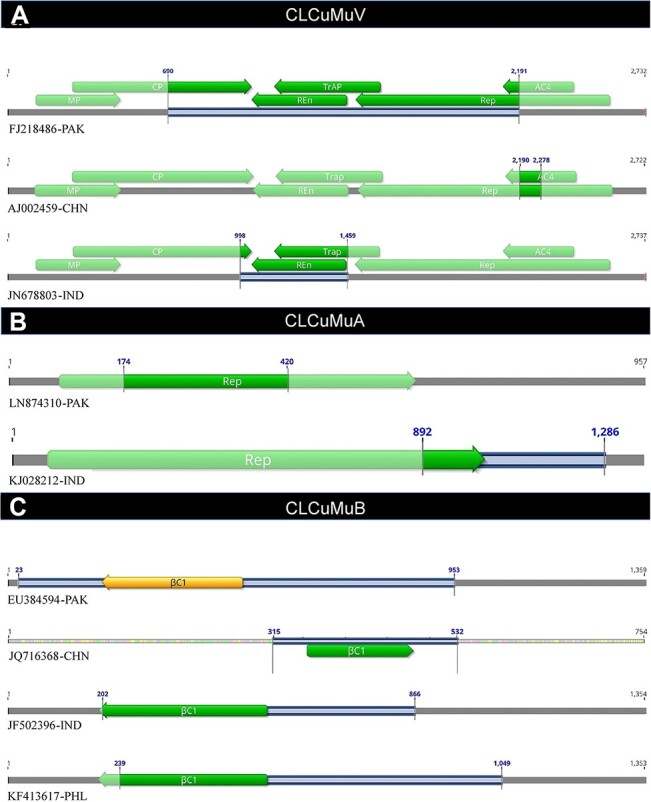
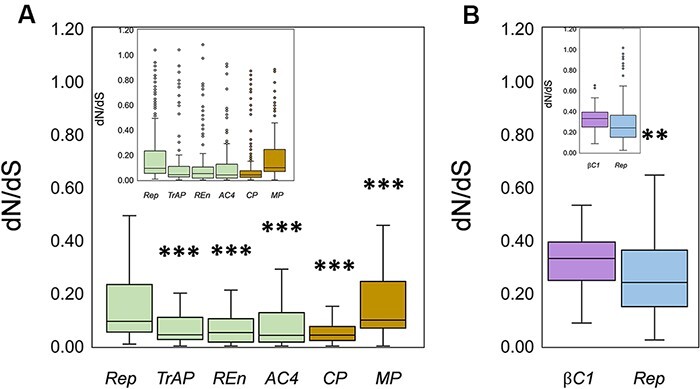
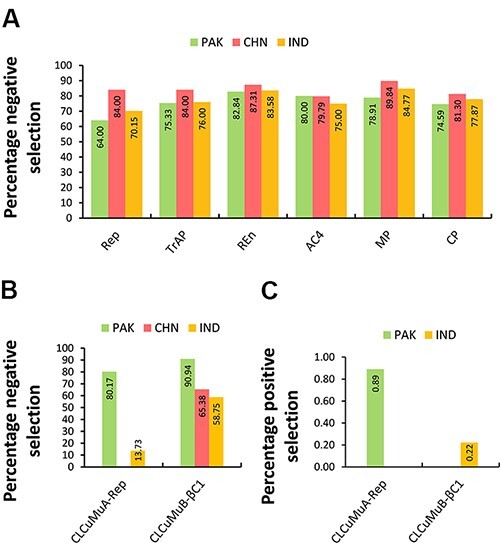
(CLCuMuV) and its associated satellites are a major part of the cotton leaf curl disease (CLCuD) caused by the begomovirus species complex. Despite the implementation of potential disease management strategies, the incessant resurgence of resistance-breaking variants of CLCuMuV imposes a continuous threat to cotton production. Here, we present a focused effort to map the geographical prevalence, genomic diversity, and molecular evolutionary endpoints that enhance disease complexity by facilitating the successful adaptation of CLCuMuV populations to the diversified ecosystems. Our results demonstrate that CLCuMuV populations are predominantly distributed in China, while the majority of alphasatellites and betasatellites exist in Pakistan. We demonstrate that together with frequent recombination, an uneven genetic variation mainly drives CLCuMuV and its satellite's virulence and evolvability. However, the pattern and distribution of recombination breakpoints greatly vary among viral and satellite sequences. The CLCuMuV, , and populations arising from distinct regions exhibit high mutation rates. Although evolutionarily linked, these populations are independently evolving under strong purifying selection. These findings will facilitate to comprehensively understand the standing genetic variability and evolutionary patterns existing among CLCuMuV populations across major cotton-producing regions of the world.
棉花曲叶多联体病毒(CLCuMuV)及其相关卫星是由菜豆金色花叶病毒属病毒复合体引起的棉花曲叶病(CLCuD)的主要组成部分。尽管实施了潜在的病害管理策略,但CLCuMuV抗性突破变异体的不断再现对棉花生产构成持续威胁。在此,我们集中精力绘制地理流行情况、基因组多样性和分子进化终点图谱,这些通过促进CLCuMuV种群成功适应多样化生态系统而增加了病害复杂性。我们的结果表明,CLCuMuV种群主要分布在中国,而大多数α卫星和β卫星存在于巴基斯坦。我们证明,频繁重组以及不均衡的遗传变异共同主要驱动CLCuMuV及其卫星的毒力和进化能力。然而,重组断点的模式和分布在病毒和卫星序列之间有很大差异。来自不同地区的CLCuMuV种群表现出高突变率。尽管在进化上有联系,但这些种群在强烈的纯化选择下独立进化。这些发现将有助于全面了解世界主要棉花产区CLCuMuV种群中存在的现有遗传变异性和进化模式。