National Microbiology Laboratory, Public Health Agency of Canada, Guelph, Ontario, Canada.
Canadian Food Inspection Agency, Winnipeg, MB, Canada.
Microb Genom. 2021 Sep;7(9). doi: 10.1099/mgen.0.000651.
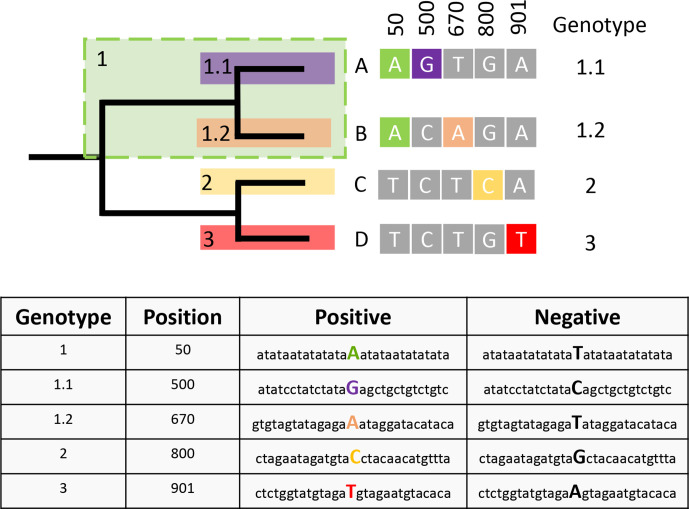
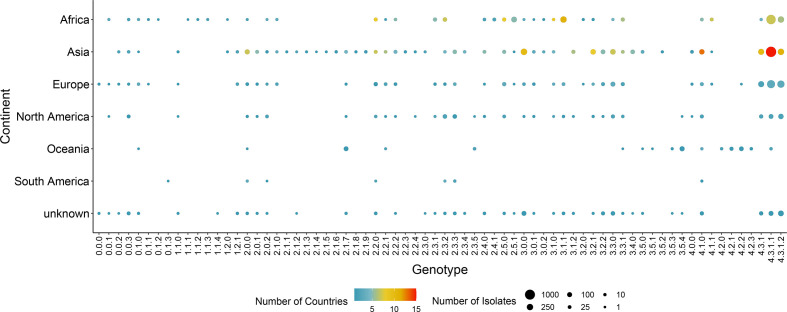
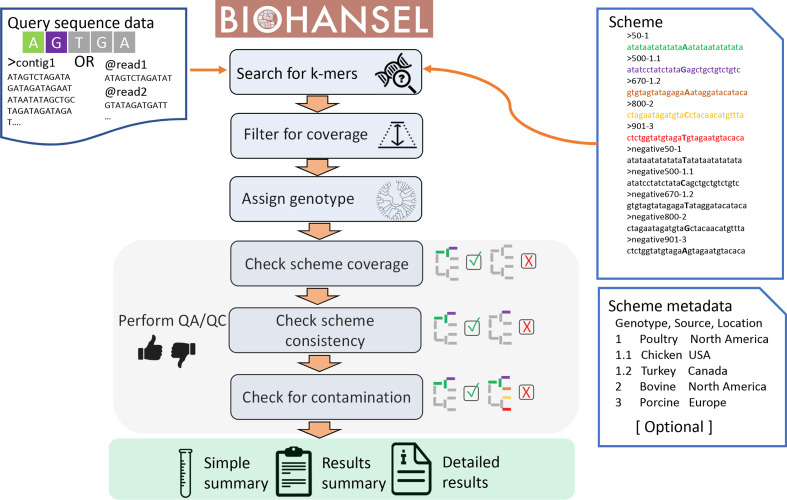
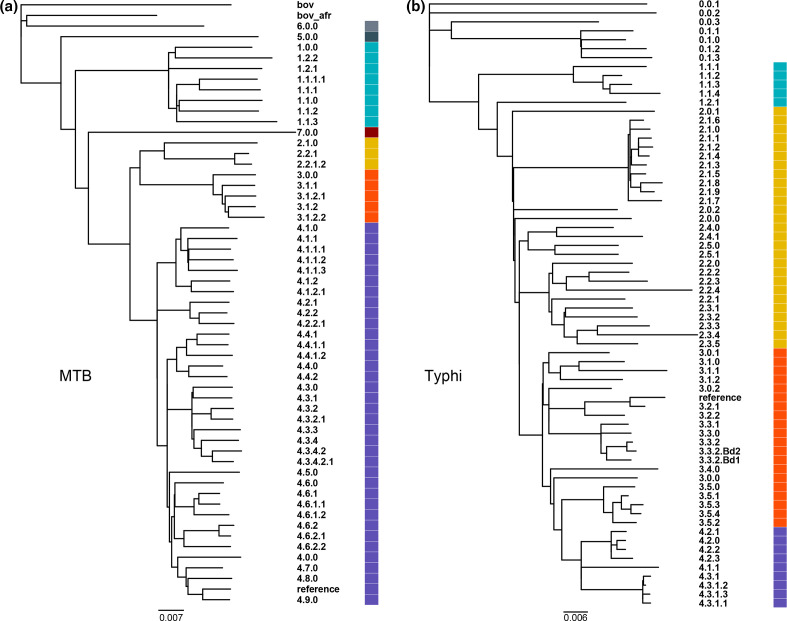
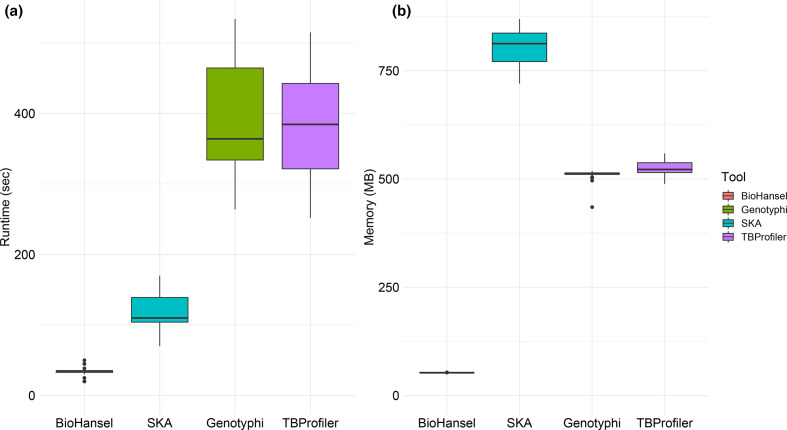
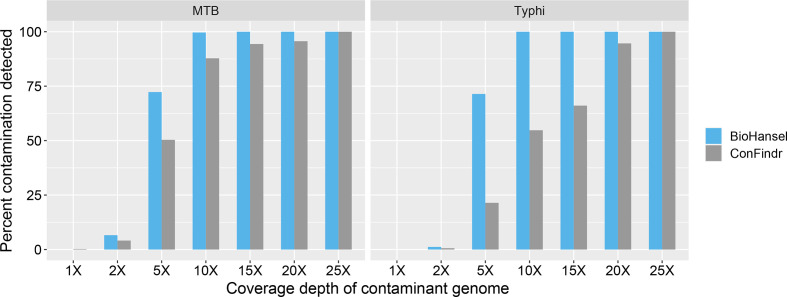
Hierarchical genotyping approaches can provide insights into the source, geography and temporal distribution of bacterial pathogens. Multiple hierarchical SNP genotyping schemes have previously been developed so that new isolates can rapidly be placed within pre-computed population structures, without the need to rebuild phylogenetic trees for the entire dataset. This classification approach has, however, seen limited uptake in routine public health settings due to analytical complexity and the lack of standardized tools that provide clear and easy ways to interpret results. The BioHansel tool was developed to provide an organism-agnostic tool for hierarchical SNP-based genotyping. The tool identifies split k-mers that distinguish predefined lineages in whole genome sequencing (WGS) data using SNP-based genotyping schemes. BioHansel uses the Aho-Corasick algorithm to type isolates from assembled genomes or raw read sequence data in a matter of seconds, with limited computational resources. This makes BioHansel ideal for use by public health agencies that rely on WGS methods for surveillance of bacterial pathogens. Genotyping results are evaluated using a quality assurance module which identifies problematic samples, such as low-quality or contaminated datasets. Using existing hierarchical SNP schemes for and Typhi, we compare the genotyping results obtained with the k-mer-based tools BioHansel and SKA, with those of the organism-specific tools TBProfiler and genotyphi, which use gold-standard reference-mapping approaches. We show that the genotyping results are fully concordant across these different methods, and that the k-mer-based tools are significantly faster. We also test the ability of the BioHansel quality assurance module to detect intra-lineage contamination and demonstrate that it is effective, even in populations with low genetic diversity. We demonstrate the scalability of the tool using a dataset of ~8100 . Typhi public genomes and provide the aggregated results of geographical distributions as part of the tool's output. BioHansel is an open source Python 3 application available on PyPI and Conda repositories and as a Galaxy tool from the public Galaxy Toolshed. In a public health context, BioHansel enables rapid and high-resolution classification of bacterial pathogens with low genetic diversity.
分层基因分型方法可以深入了解细菌病原体的来源、地理位置和时间分布。以前已经开发了多种分层 SNP 基因分型方案,以便可以快速将新分离株放置在预先计算的种群结构中,而无需为整个数据集重建系统发育树。然而,由于分析复杂性以及缺乏提供清晰易用的解释结果的标准化工具,这种分类方法在常规公共卫生环境中的应用受到限制。BioHansel 工具是为基于 SNP 的分层基因分型开发的一种与生物体无关的工具。该工具使用基于 SNP 的基因分型方案,使用 SNP 识别区分全基因组测序 (WGS) 数据中预定义谱系的分裂 k-mer。BioHansel 使用 Aho-Corasick 算法在几秒钟内对组装基因组或原始读取序列数据中的分离株进行分型,所需的计算资源有限。这使得 BioHansel 非常适合依赖 WGS 方法进行细菌病原体监测的公共卫生机构使用。使用质量保证模块评估基因分型结果,该模块可识别有问题的样本,例如低质量或污染的数据集。我们使用 和 沙门氏菌的现有分层 SNP 方案,将基于 k-mer 的工具 BioHansel 和 SKA 与专门针对生物体的工具 TBProfiler 和 genotyphi 获得的基因分型结果进行比较,后者使用黄金标准参考映射方法。我们表明,这些不同方法的基因分型结果完全一致,并且基于 k-mer 的工具速度明显更快。我们还测试了 BioHansel 质量保证模块检测谱系内污染的能力,并证明即使在遗传多样性低的群体中,它也是有效的。我们使用约 8100 个 伤寒沙门氏菌公共基因组数据集来测试工具的可扩展性,并提供工具输出的地理分布的聚合结果。BioHansel 是一个可在 PyPI 和 Conda 存储库中获得的开源 Python 3 应用程序,并且是公共 Galaxy Toolshed 中的 Galaxy 工具。在公共卫生环境中,BioHansel 可以快速实现具有低遗传多样性的细菌病原体的高分辨率分类。