Zhao Yajuan, Zhang Junli, Wang Shuhan, Jiang Qianqian, Xu Keshu
Division of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Front Cell Dev Biol. 2021 Sep 7;9:731790. doi: 10.3389/fcell.2021.731790. eCollection 2021.
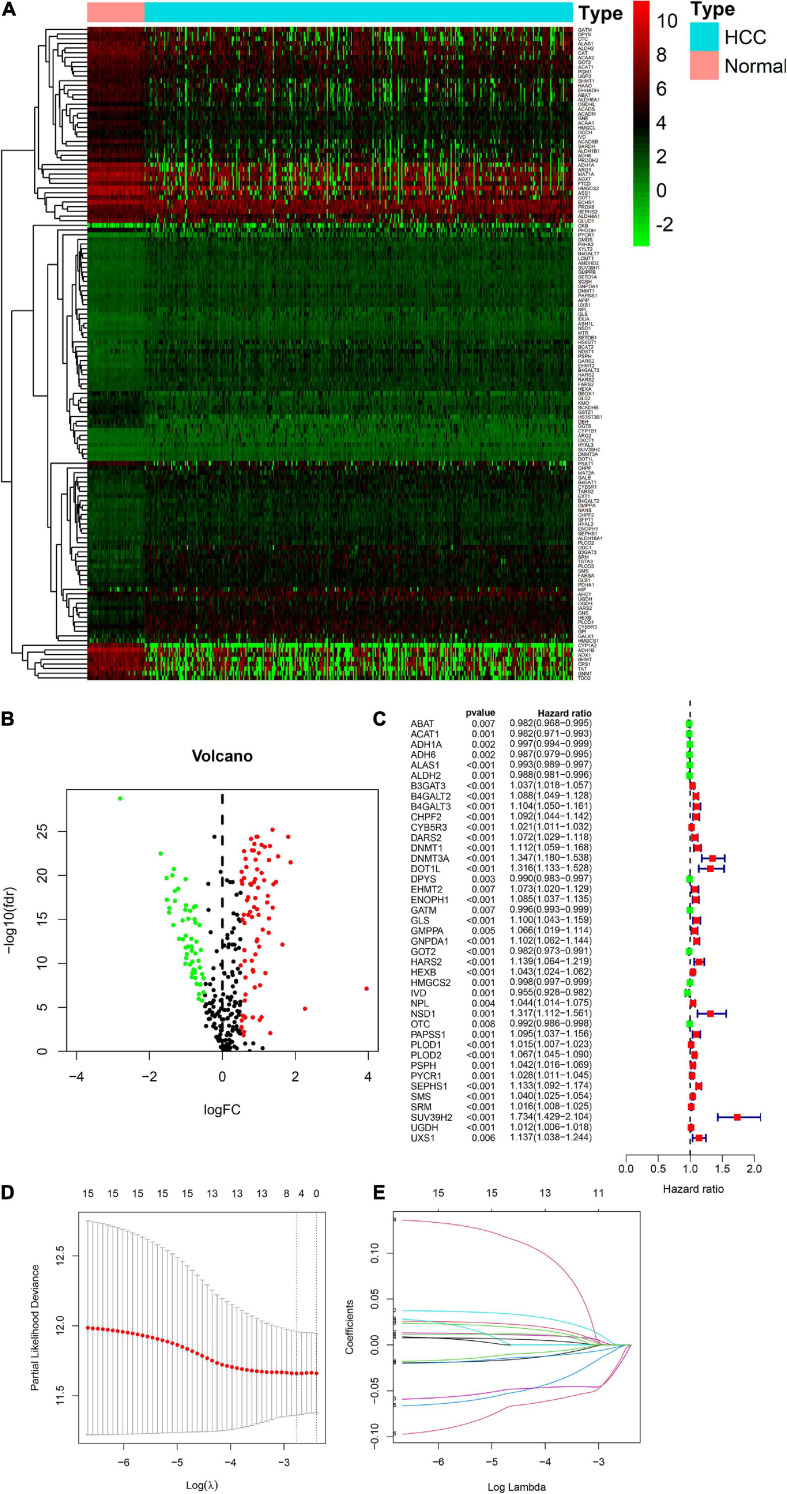
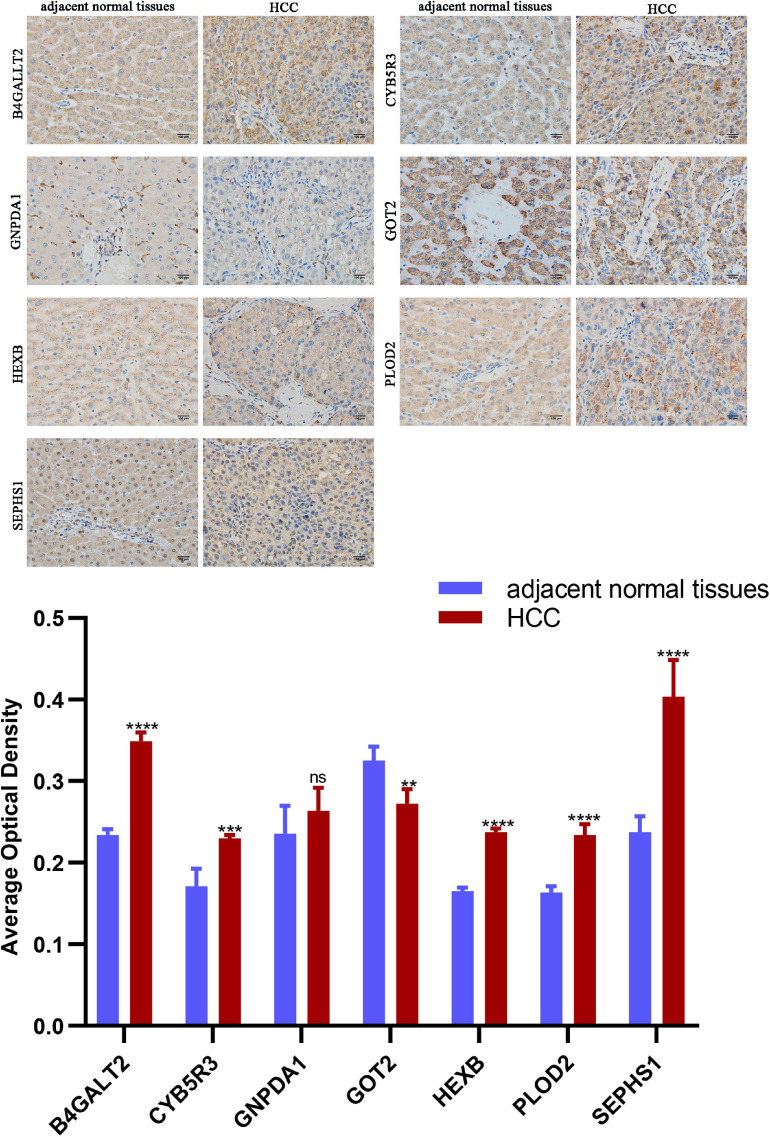
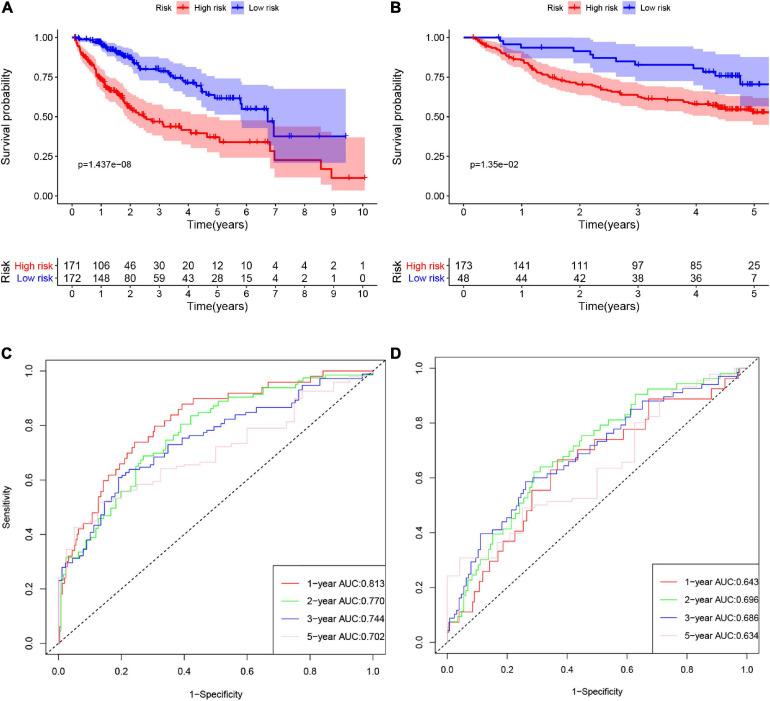
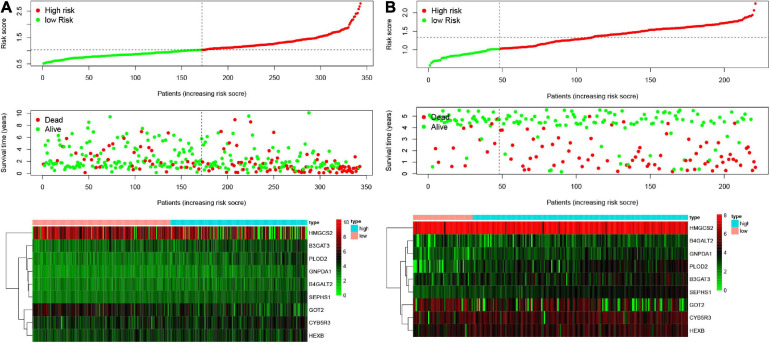
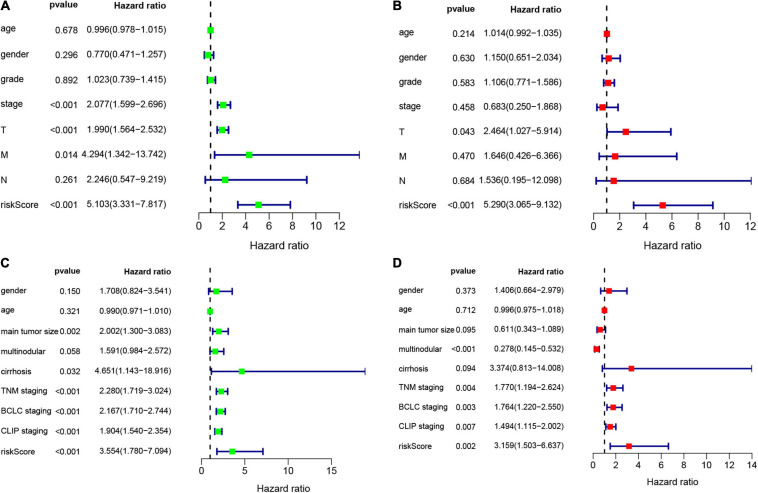
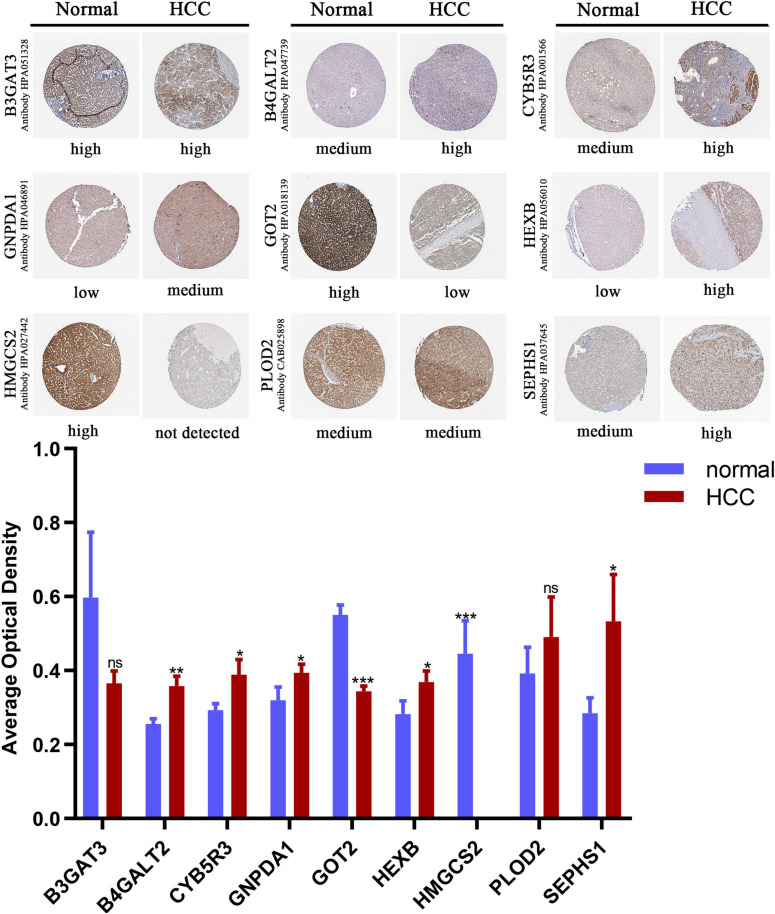
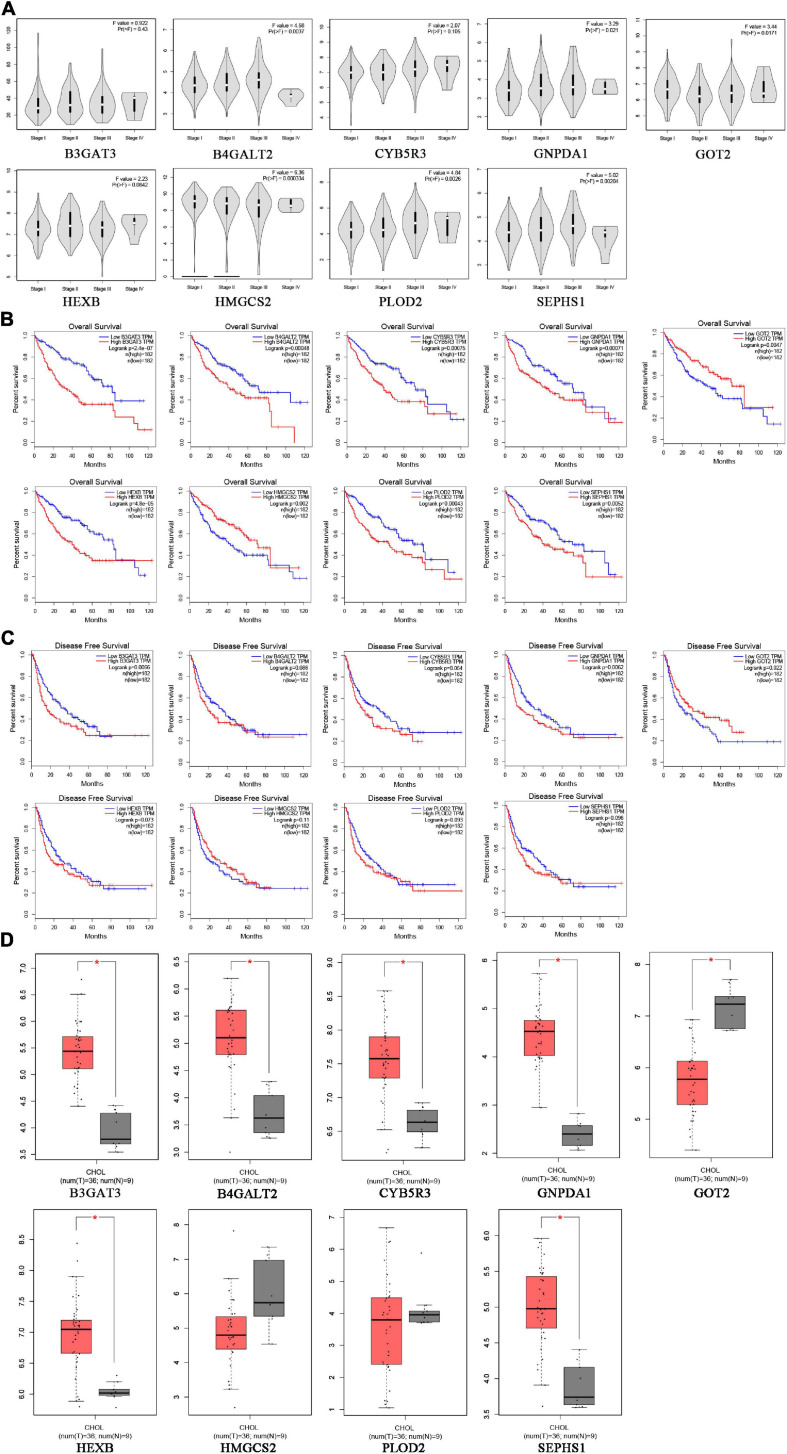
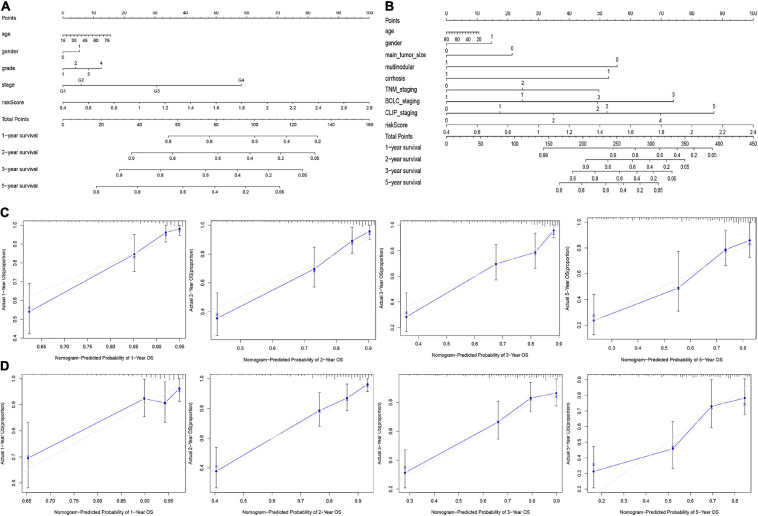
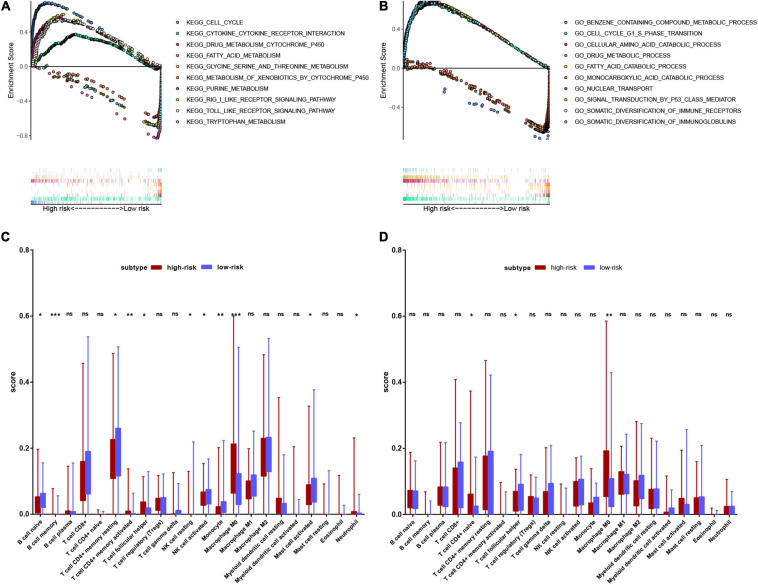
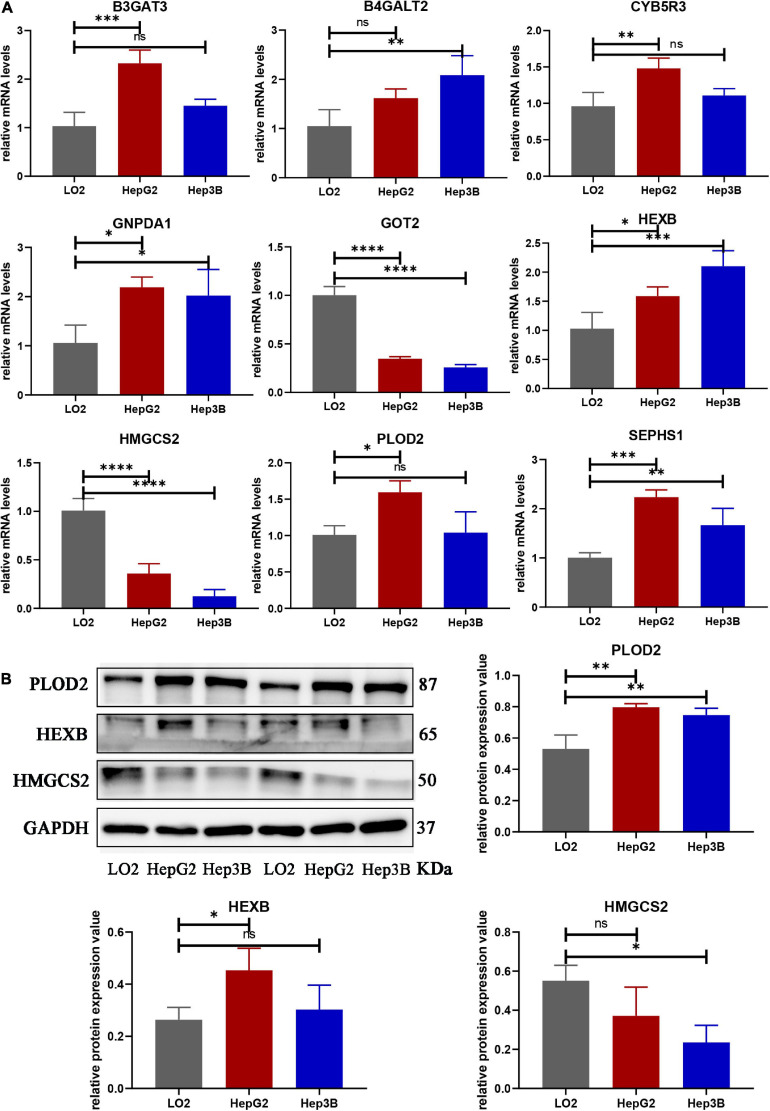
Hepatocellular carcinoma (HCC) is the world's second most deadly cancer, and metabolic reprogramming is its distinguishing feature. Among metabolite profiling, variation in amino acid metabolism supports tumor proliferation and metastasis to the most extent, yet a systematic study on the role of amino acid metabolism-related genes in HCC is still lacking. An effective amino acid metabolism-related prediction signature is urgently needed to assess the prognosis of HCC patients for individualized treatment. RNA-seq data of HCC from the TCGA-LIHC and GSE14520 (GPL3921) datasets were defined as the training set and validation set, respectively. Amino acid metabolic genes were extracted from the Molecular Signature Database. Univariate Cox and LASSO regression analyses were performed to build a predictive risk signature. K-M curves, ROC curves, and univariate and multivariate Cox regression were conducted to evaluate the predictive value of this risk signature. Functional enrichment was analyzed by GSEA and CIBERSORTx software. A nine-gene amino acid metabolism-related risk signature including B3GAT3, B4GALT2, CYB5R3, GNPDA1, GOT2, HEXB, HMGCS2, PLOD2, and SEPHS1 was constructed to predict the overall survival (OS) of HCC patients. Patients were separated into high-risk and low-risk groups based on risk scores and low-risk patients had lower risk scores and longer survival time. Univariate and multivariate Cox regression verified that this signature was an independent risk factor for HCC. ROC curves showed that this risk signature can effectively predict the 1-, 2-, 3- and 5-year survival times of patients with HCC. Additionally, prognostic nomograms were established based on the training set and validation set. These genes were closely correlated with the immune regulation. Our study identified a nine-gene amino acid metabolism-related risk signature and built predictive nomograms for OS in HCC. These findings will help us to personalize the treatment of liver cancer patients.
肝细胞癌(HCC)是全球第二大致命癌症,代谢重编程是其显著特征。在代谢物谱分析中,氨基酸代谢的变化在最大程度上支持肿瘤增殖和转移,但目前仍缺乏关于氨基酸代谢相关基因在HCC中作用的系统性研究。迫切需要一个有效的氨基酸代谢相关预测特征来评估HCC患者的预后,以实现个体化治疗。来自TCGA-LIHC和GSE14520(GPL3921)数据集的HCC的RNA测序数据分别被定义为训练集和验证集。从分子特征数据库中提取氨基酸代谢基因。进行单变量Cox和LASSO回归分析以构建预测风险特征。绘制K-M曲线、ROC曲线,并进行单变量和多变量Cox回归以评估该风险特征的预测价值。通过GSEA和CIBERSORTx软件进行功能富集分析。构建了一个包含B3GAT3、B4GALT2、CYB5R3、GNPDA1、GOT2、HEXB、HMGCS2、PLOD2和SEPHS1的九基因氨基酸代谢相关风险特征,用于预测HCC患者的总生存期(OS)。根据风险评分将患者分为高风险组和低风险组,低风险患者的风险评分较低且生存时间更长。单变量和多变量Cox回归验证了该特征是HCC的独立危险因素。ROC曲线表明,该风险特征可以有效预测HCC患者1年、2年、3年和5年的生存时间。此外,基于训练集和验证集建立了预后列线图。这些基因与免疫调节密切相关。我们的研究确定了一个九基因氨基酸代谢相关风险特征,并构建了HCC患者OS的预测列线图。这些发现将有助于我们实现肝癌患者的个体化治疗。