Rahalkar Hasumati, Sheppard Alan, Santos Gustavo Mendes Lima, Dasgupta Chitralekha, Perez-Tapia Sonia Mayra, Lopez-Morales Carlos A, Salek Sam
Metina PharmConsulting Pvt. Ltd., Navi Mumbai, India.
School of Life and Medical Sciences, University of Hertfordshire, Hatfield, United Kingdom.
Front Med (Lausanne). 2021 Sep 9;8:726660. doi: 10.3389/fmed.2021.726660. eCollection 2021.
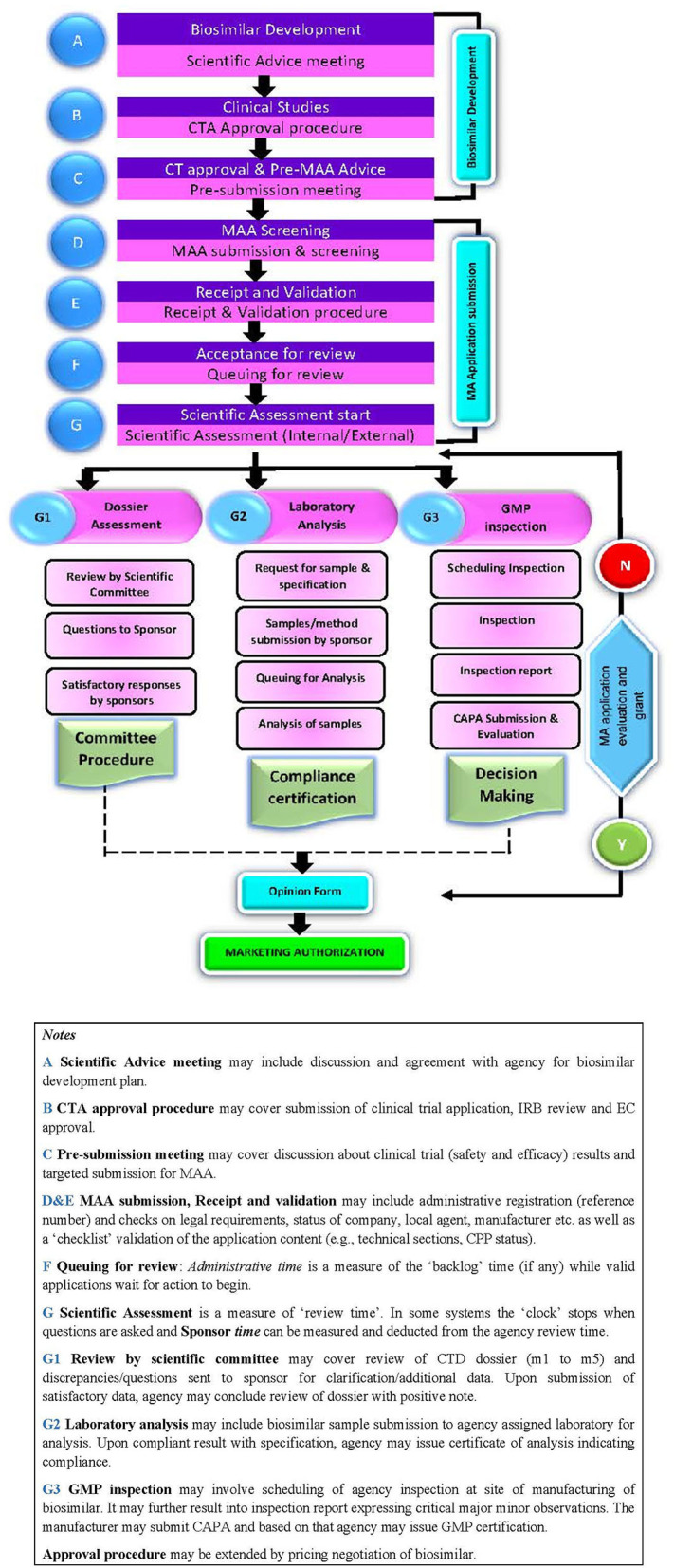
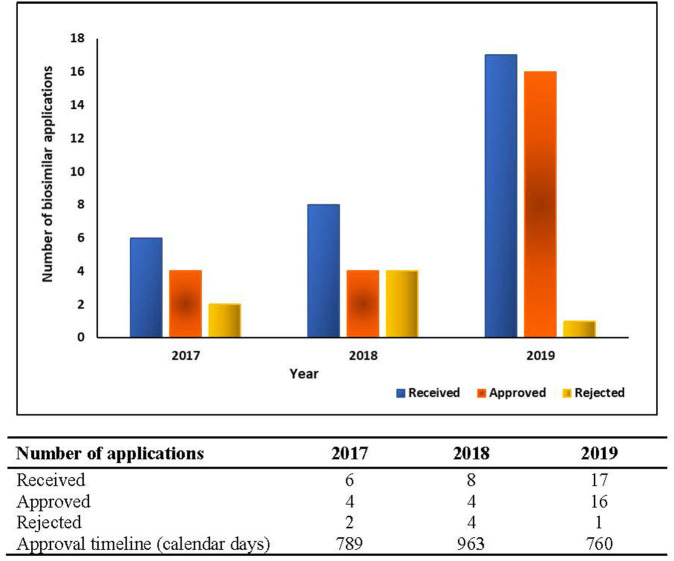
The aim of the study was to identify, interpret, and compare the current perspectives of regulatory agencies in six member countries of BRICS-TM (Brazil, Russia, India, China, South Africa, Turkey, and Mexico) on the different criteria used for biosimilar development and marketing authorisation process. A semi-quantitative questionnaire was developed covering the organisation of agency, biosimilar development criteria and marketing authorisation process and sent to seven regulatory agencies covering the BRICS-TM countries. All data was kept anonymous and confidential. Data processing and analysis was carried out; descriptive statistics were used for quantitative data and content analysis was employed to generate themes for qualitative data. Out of the seven regulatory agencies included in the study, six representatives provided the responses. The perspectives of these six regulatory agencies varied on a number of aspects relating to the review criteria for biosimilar development and licencing process. The most prevalent model for data assessment is the "full review" of a marketing authorisation application. There is lack of a standard approach across the agencies on sourcing of the reference biological product, toxicity studies and confirmatory clinical studies. Most agencies restrict interaction with biosimilar developers and any scientific advice is non-binding. The marketing authorisation approval depends on scientific assessment of the dossier, sample analysis and GMP certification. The agencies do not issue any public assessment report specifying the summary basis of biosimilar approval. Regulatory agencies across the six emerging economies are steadily improving the regulatory mechanism in the area of biosimilars. However, there remains scope for increasing the effectiveness and efficiency of the processes by encouraging open and transparent interaction with developers, adopting a flexible approach toward accepting advanced analytical data in lieu of clinical studies and enhancing regulatory reliance amongst agencies. This will help to simplify the new biosimilar development programmes and make them more cost-effective.
该研究的目的是识别、解读并比较金砖五国-土耳其-墨西哥(巴西、俄罗斯、印度、中国、南非、土耳其和墨西哥)六个成员国监管机构对生物类似药研发及上市许可过程中使用的不同标准的当前观点。为此制定了一份半定量问卷,内容涵盖机构组织、生物类似药研发标准及上市许可流程,并发送给了涵盖金砖五国-土耳其-墨西哥国家的七个监管机构。所有数据均保持匿名和保密。进行了数据处理与分析;定量数据采用描述性统计,定性数据采用内容分析以生成主题。在该研究纳入的七个监管机构中,有六个代表提供了回复。这六个监管机构在生物类似药研发及许可流程的审查标准的多个方面观点各异。数据评估最普遍的模式是对上市许可申请进行“全面审查”。各机构在参照生物制品的来源、毒性研究和确证性临床研究方面缺乏标准方法。大多数机构限制与生物类似药开发者的互动,任何科学建议均无约束力。上市许可审批取决于对档案的科学评估、样品分析和GMP认证。各机构不发布任何具体说明生物类似药批准简要依据的公开评估报告。六个新兴经济体的监管机构在生物类似药领域的监管机制正在稳步改善。然而,通过鼓励与开发者进行开放透明的互动、采取灵活方式接受先进分析数据以替代临床研究以及增强各机构间的监管互信,仍有提高流程有效性和效率的空间。这将有助于简化新的生物类似药研发项目并使其更具成本效益。