Department of Biology, The Bioinformatics Centre, University of Copenhagen, Copenhagen, Denmark.
BMC Bioinformatics. 2021 Sep 29;22(1):470. doi: 10.1186/s12859-021-04375-2.
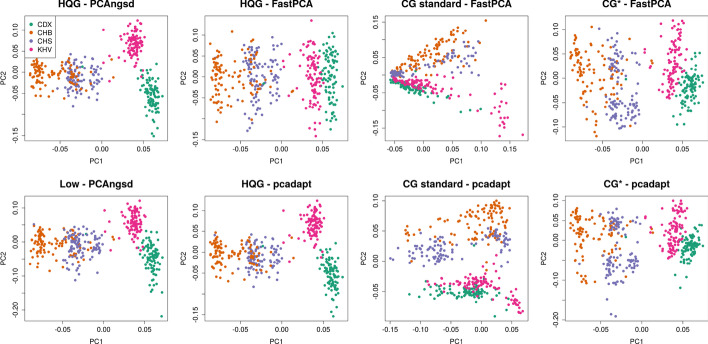
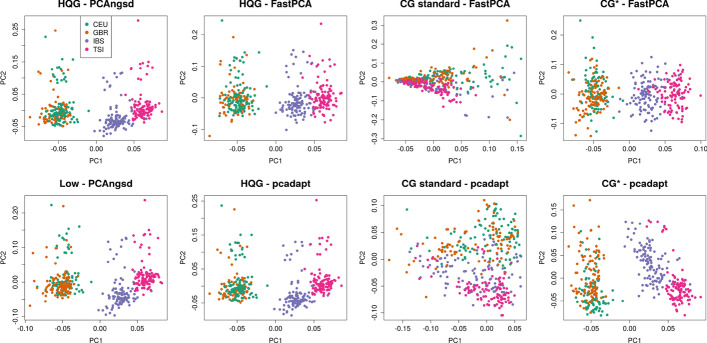
Identification of selection signatures between populations is often an important part of a population genetic study. Leveraging high-throughput DNA sequencing larger sample sizes of populations with similar ancestries has become increasingly common. This has led to the need of methods capable of identifying signals of selection in populations with a continuous cline of genetic differentiation. Individuals from continuous populations are inherently challenging to group into meaningful units which is why existing methods rely on principal components analysis for inference of the selection signals. These existing methods require called genotypes as input which is problematic for studies based on low-coverage sequencing data.
We have extended two principal component analysis based selection statistics to genotype likelihood data and applied them to low-coverage sequencing data from the 1000 Genomes Project for populations with European and East Asian ancestry to detect signals of selection in samples with continuous population structure.
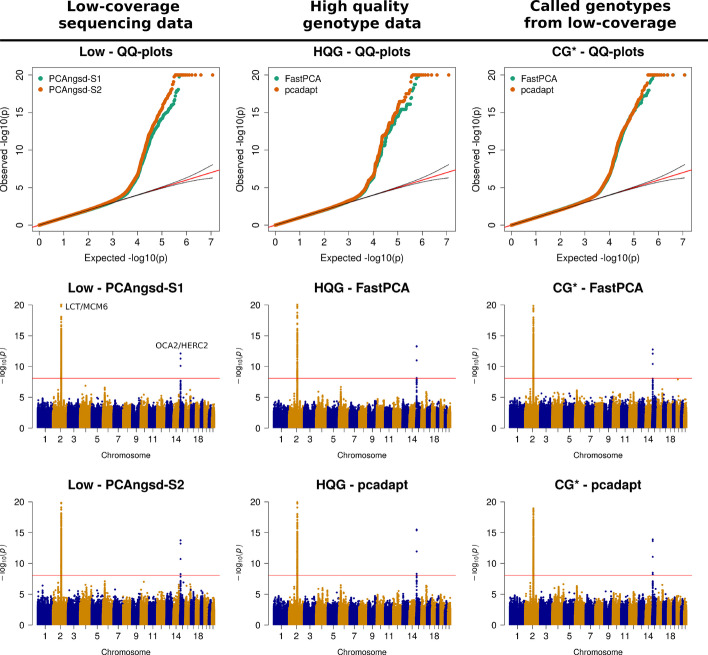
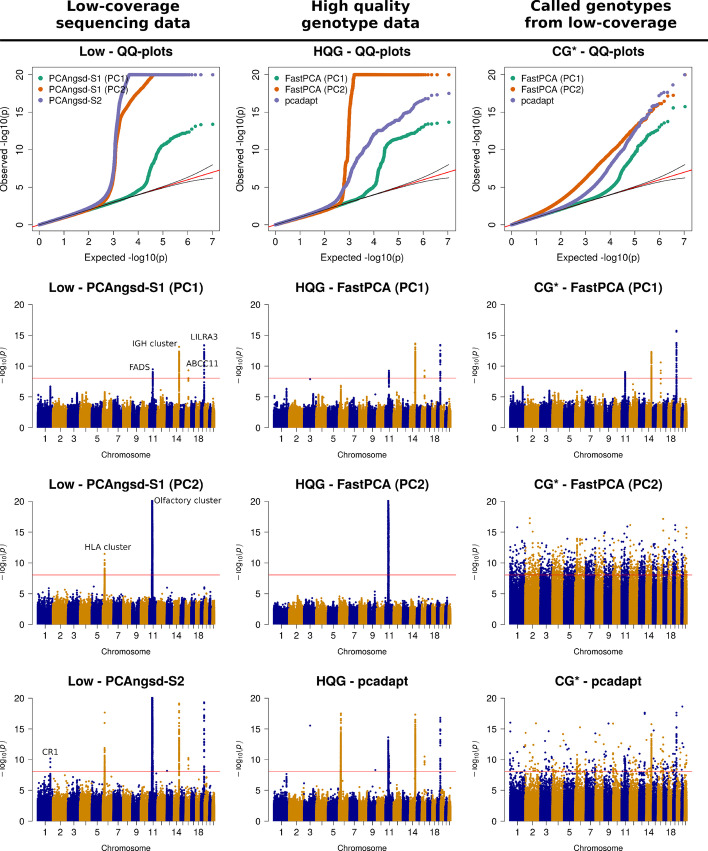
Here, we present two selections statistics which we have implemented in the PCAngsd framework. These methods account for genotype uncertainty, opening for the opportunity to conduct selection scans in continuous populations from low and/or variable coverage sequencing data. To illustrate their use, we applied the methods to low-coverage sequencing data from human populations of East Asian and European ancestries and show that the implemented selection statistics can control the false positive rate and that they identify the same signatures of selection from low-coverage sequencing data as state-of-the-art software using high quality called genotypes.
We show that selection scans of low-coverage sequencing data of populations with similar ancestry perform on par with that obtained from high quality genotype data. Moreover, we demonstrate that PCAngsd outperform selection statistics obtained from called genotypes from low-coverage sequencing data without the need for ad-hoc filtering.
在群体间识别选择信号通常是群体遗传学研究的重要组成部分。利用高通量 DNA 测序,对具有相似祖先的较大样本群体进行研究变得越来越普遍。这导致需要能够识别具有连续遗传分化渐变的群体中选择信号的方法。连续群体中的个体本身就难以分组为有意义的单位,这就是为什么现有的方法依赖于主成分分析来推断选择信号的原因。这些现有的方法需要输入已调用的基因型,这对于基于低覆盖测序数据的研究来说是有问题的。
我们已经将两种基于主成分分析的选择统计方法扩展到基因型似然数据,并将其应用于来自 1000 基因组计划的具有欧洲和东亚祖先的连续群体的低覆盖测序数据中,以检测具有连续群体结构的样本中的选择信号。
在这里,我们提出了两种选择统计方法,我们已经在 PCAngsd 框架中实现了这些方法。这些方法考虑了基因型的不确定性,为在低覆盖度和/或可变覆盖度测序数据的连续群体中进行选择扫描提供了机会。为了说明它们的用途,我们将这些方法应用于东亚和欧洲祖先人群的低覆盖测序数据,并表明所实现的选择统计方法可以控制假阳性率,并且它们可以从低覆盖测序数据中识别与使用高质量已调用基因型的最新软件相同的选择信号。
我们表明,具有相似祖先的低覆盖测序数据的选择扫描与从高质量基因型数据获得的扫描相当。此外,我们证明了 PCAngsd 优于从低覆盖测序数据的已调用基因型获得的选择统计数据,而无需进行特殊过滤。