Department of Computer Science, University of Illinois at Urbana-Champaign, Urbana, Illinois, United States of America.
PLoS Comput Biol. 2021 Oct 6;17(10):e1008950. doi: 10.1371/journal.pcbi.1008950. eCollection 2021 Oct.
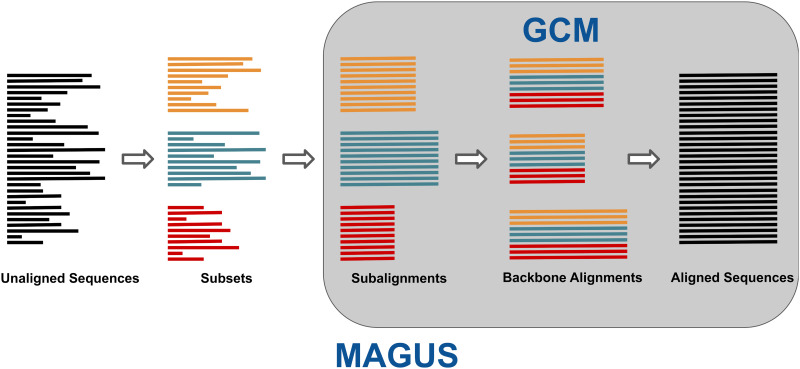
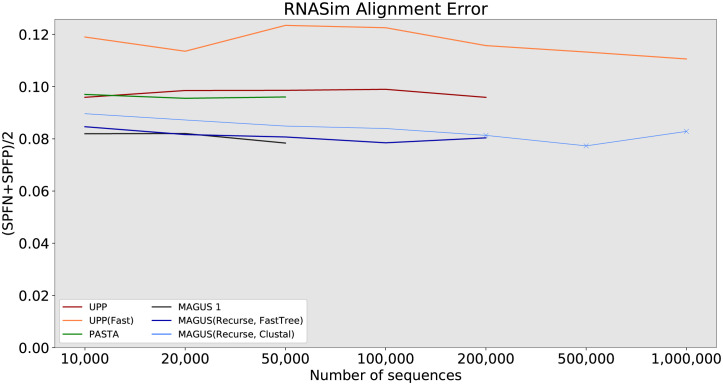
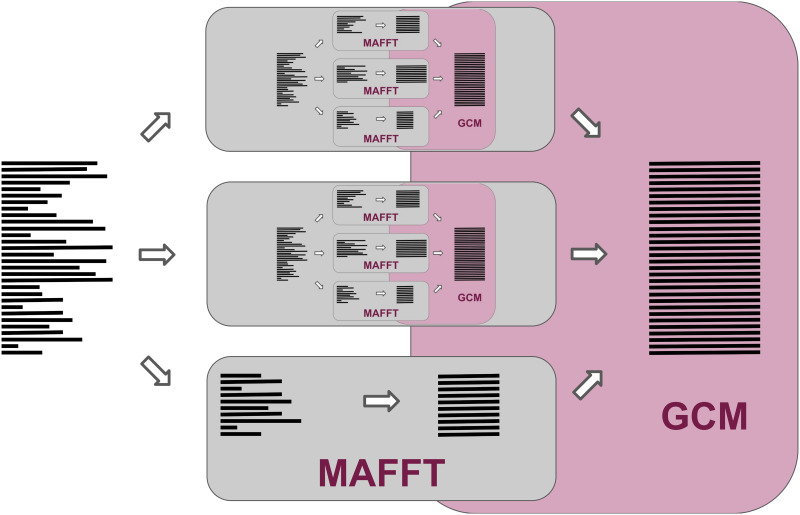
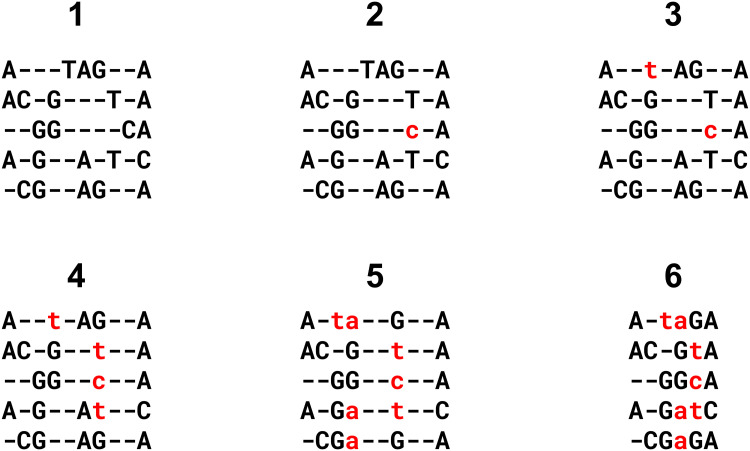
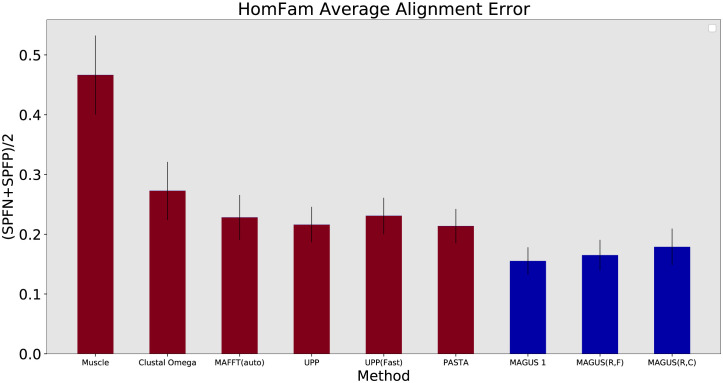
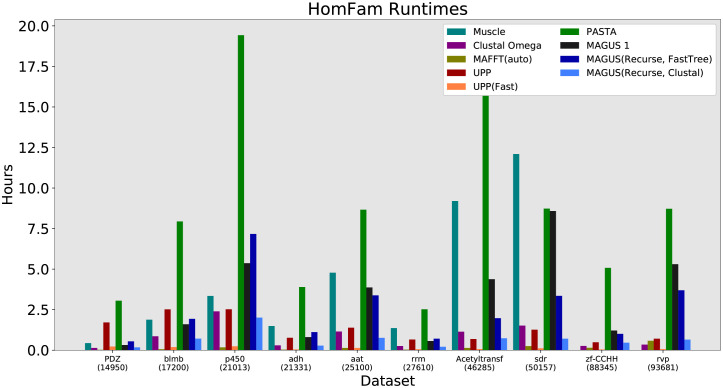
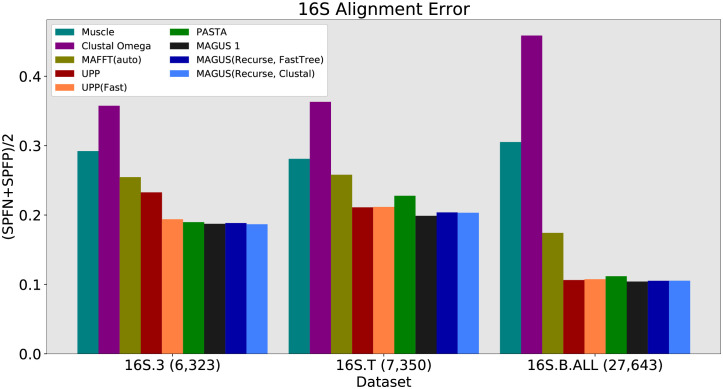
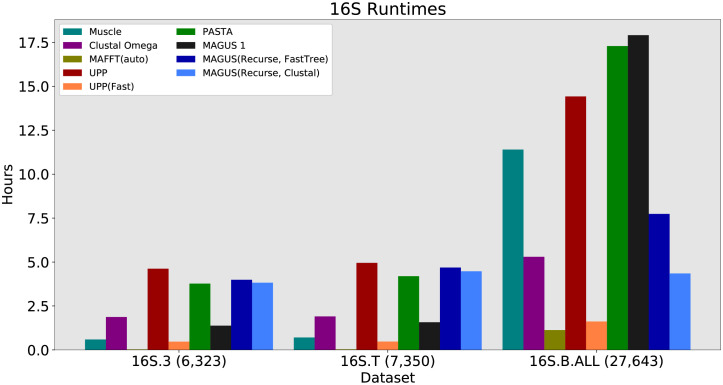
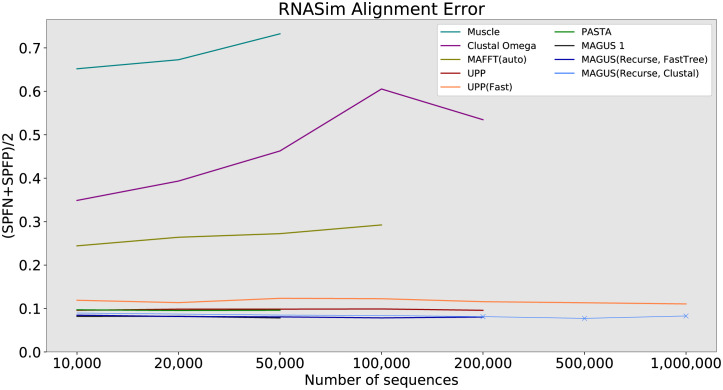
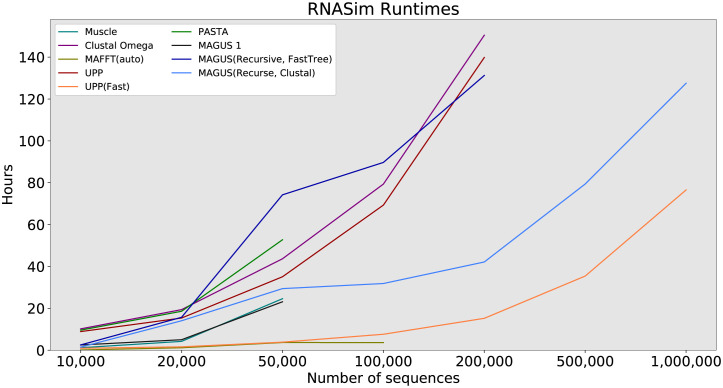
Multiple sequence alignment tools struggle to keep pace with rapidly growing sequence data, as few methods can handle large datasets while maintaining alignment accuracy. We recently introduced MAGUS, a new state-of-the-art method for aligning large numbers of sequences. In this paper, we present a comprehensive set of enhancements that allow MAGUS to align vastly larger datasets with greater speed. We compare MAGUS to other leading alignment methods on datasets of up to one million sequences. Our results demonstrate the advantages of MAGUS over other alignment software in both accuracy and speed. MAGUS is freely available in open-source form at https://github.com/vlasmirnov/MAGUS.
多序列比对工具难以跟上快速增长的序列数据,因为很少有方法可以在保持比对准确性的同时处理大型数据集。我们最近引入了 MAGUS,这是一种用于对齐大量序列的最新方法。在本文中,我们提出了一组全面的增强功能,使 MAGUS 能够以更快的速度对齐更大的数据集。我们在多达一百万条序列的数据集上将 MAGUS 与其他领先的比对方法进行了比较。我们的结果表明,MAGUS 在准确性和速度方面都优于其他比对软件。MAGUS 可在 https://github.com/vlasmirnov/MAGUS 上以开源形式免费获得。