Lüth Theresa, Wasner Kobi, Klein Christine, Schaake Susen, Tse Ronnie, Pereira Sandro L, Laß Joshua, Sinkkonen Lasse, Grünewald Anne, Trinh Joanne
Institute of Neurogenetics BMF, University of Lübeck and University Hospital Schleswig-Holstein Campus Lübeck, Lübeck, Germany.
Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Belvaux, Luxembourg.
Front Aging Neurosci. 2021 Sep 28;13:713084. doi: 10.3389/fnagi.2021.713084. eCollection 2021.
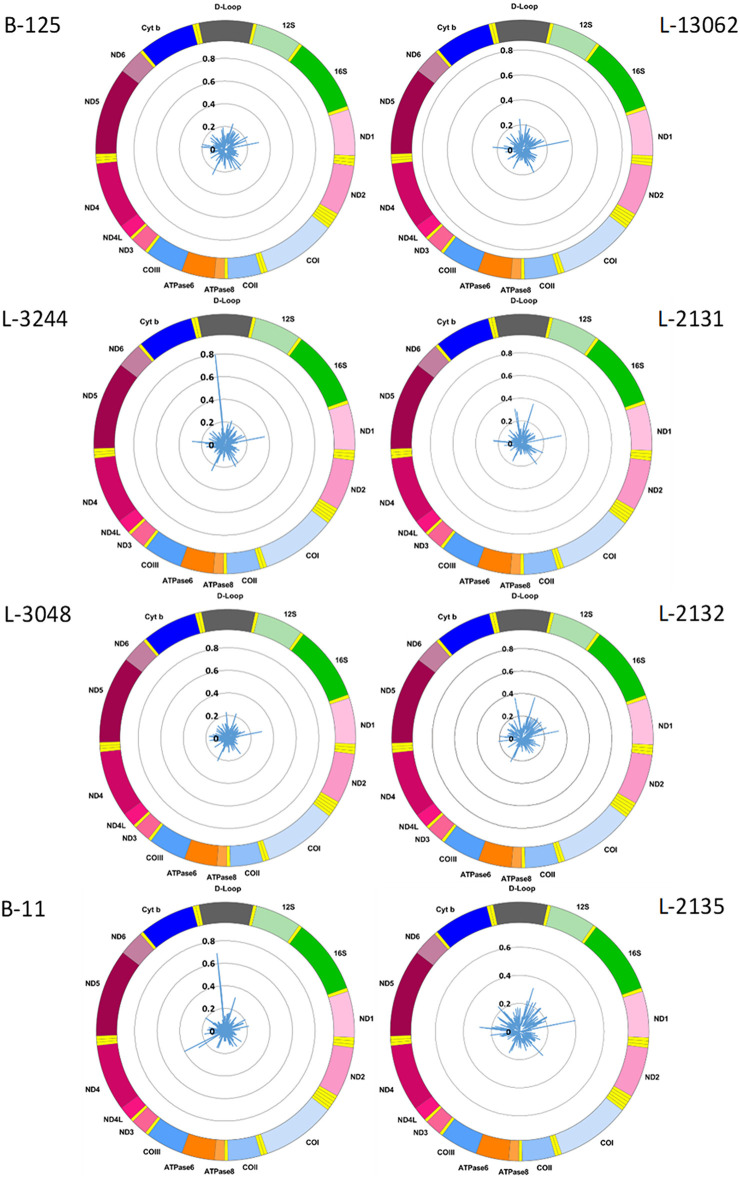
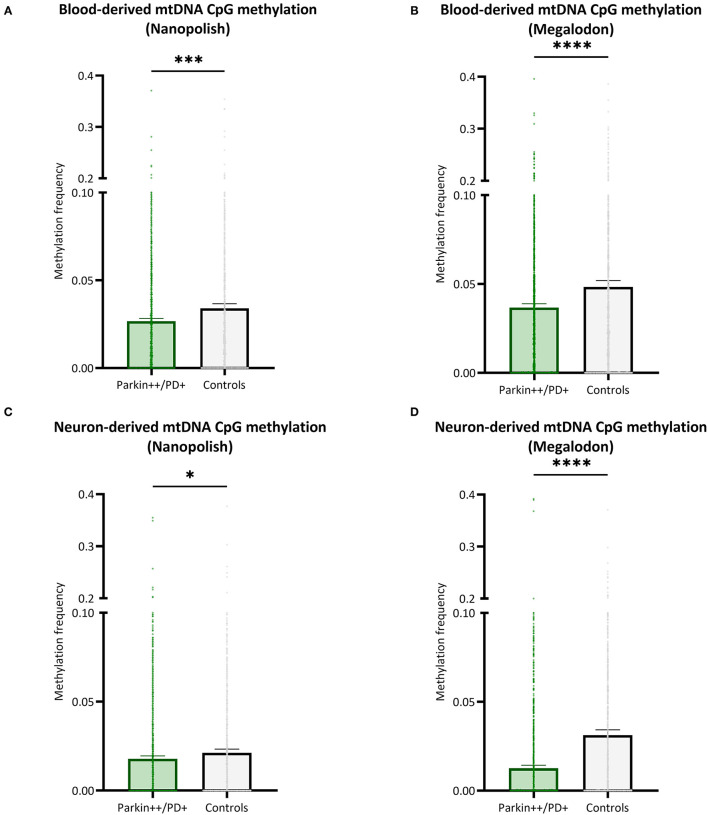
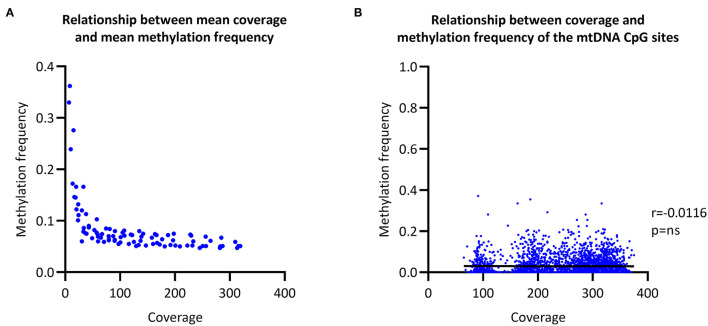
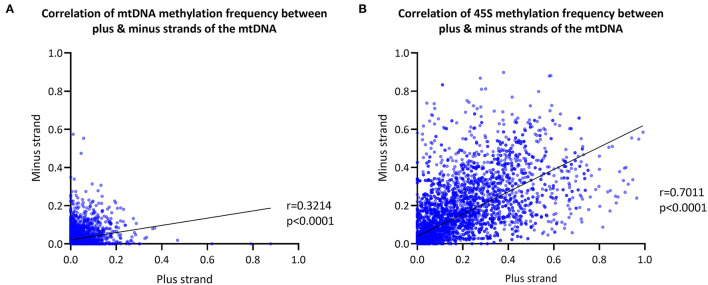
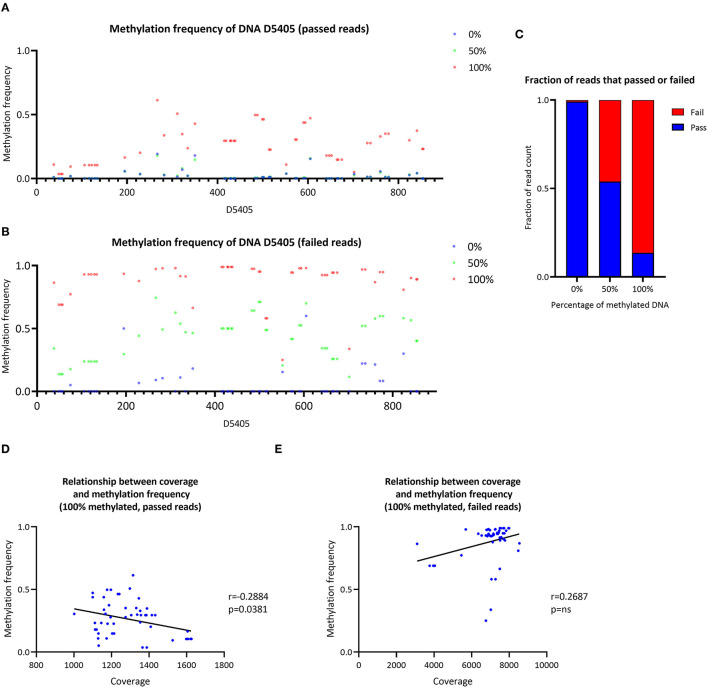
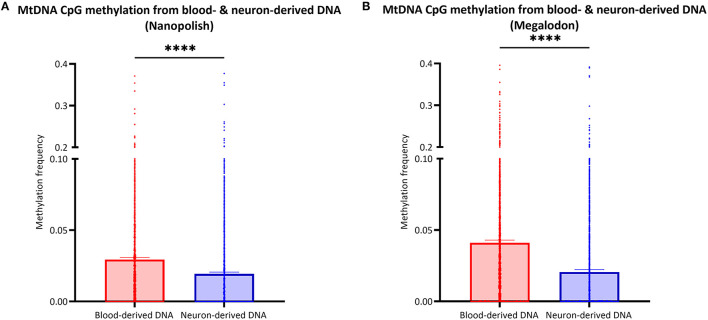
To establish a workflow for mitochondrial DNA (mtDNA) CpG methylation using Nanopore whole-genome sequencing and perform first pilot experiments on affected biallelic mutation carriers (Parkin-PD) and healthy controls. Mitochondria, including mtDNA, are established key players in Parkinson's disease (PD) pathogenesis. Mutations in Parkin, essential for degradation of damaged mitochondria, cause early-onset PD. However, mtDNA methylation and its implication in PD is understudied. Herein, we establish a workflow using Nanopore sequencing to directly detect mtDNA CpG methylation and compare mtDNA methylation between Parkin-related PD and healthy individuals. To obtain mtDNA, whole-genome Nanopore sequencing was performed on blood-derived from five Parkin-PD and three control subjects. In addition, induced pluripotent stem cell (iPSC)-derived midbrain neurons from four of these patients with PD and the three control subjects were investigated. The workflow was validated, using methylated and unmethylated 897 bp synthetic DNA samples at different dilution ratios (0, 50, 100% methylation) and mtDNA without methylation. MtDNA CpG methylation frequency (MF) was detected using Nanopolish and Megalodon. Across all blood-derived samples, we obtained a mean coverage of 250.3X (SD ± 80.5X) and across all neuron-derived samples 830X (SD ± 465X) of the mitochondrial genome. We detected overall low-level CpG methylation from the blood-derived DNA (mean MF ± SD = 0.029 ± 0.041) and neuron-derived DNA (mean MF ± SD = 0.019 ± 0.035). Validation of the workflow, using synthetic DNA samples showed that highly methylated DNA molecules were prone to lower Guppy Phred quality scores and thereby more likely to fail Guppy base-calling. CpG methylation in blood- and neuron-derived DNA was significantly lower in Parkin-PD compared to controls (Mann-Whitney U-test < 0.05). Nanopore sequencing is a useful method to investigate mtDNA methylation architecture, including Guppy-failed reads is of importance when investigating highly methylated sites. We present a mtDNA methylation workflow and suggest methylation variability across different tissues and between Parkin-PD patients and controls as an initial model to investigate.
利用纳米孔全基因组测序建立线粒体DNA(mtDNA)CpG甲基化的工作流程,并对受影响的双等位基因突变携带者(帕金森病相关帕金森病,Parkin-PD)和健康对照进行首次试点实验。线粒体,包括mtDNA,是帕金森病(PD)发病机制中已确定的关键因素。对受损线粒体降解至关重要的Parkin基因突变会导致早发性PD。然而,mtDNA甲基化及其在PD中的意义尚未得到充分研究。在此,我们建立了一种使用纳米孔测序直接检测mtDNA CpG甲基化的工作流程,并比较了Parkin相关PD患者和健康个体之间mtDNA甲基化情况。为了获取mtDNA,对来自5名Parkin-PD患者和3名对照受试者的血液进行了全基因组纳米孔测序。此外,还对其中4名PD患者和3名对照受试者诱导多能干细胞(iPSC)来源的中脑神经元进行了研究。使用不同稀释比例(0、50、100%甲基化)的甲基化和未甲基化897 bp合成DNA样本以及未甲基化的mtDNA对该工作流程进行了验证。使用Nanopolish和Megalodon检测mtDNA CpG甲基化频率(MF)。在所有血液来源的样本中,我们获得了线粒体基因组平均250.3X(标准差±80.5X)的覆盖度,在所有神经元来源的样本中获得了830X(标准差±465X)的覆盖度。我们从血液来源的DNA(平均MF±标准差=0.029±0.041)和神经元来源的DNA(平均MF±标准差=0.019±0.035)中检测到总体低水平的CpG甲基化。使用合成DNA样本对工作流程进行验证表明,高度甲基化的DNA分子更容易获得较低的Guppy Phred质量分数,因此更有可能在Guppy碱基识别中失败。与对照组相比,Parkin-PD患者血液和神经元来源的DNA中的CpG甲基化显著降低(曼-惠特尼U检验<0.05)。纳米孔测序是研究mtDNA甲基化结构的一种有用方法,在研究高度甲基化位点时,包括Guppy识别失败的读数很重要。我们展示了一种mtDNA甲基化工作流程,并提出不同组织之间以及Parkin-PD患者与对照之间的甲基化变异性作为一个初步模型以供研究。