Wake Forest School of Medicine, Winston-Salem, USA.
The University of Southern Denmark, Odense, Denmark.
BioDrugs. 2021 Nov;35(6):735-748. doi: 10.1007/s40259-021-00502-w. Epub 2021 Oct 16.
AVT02 (adalimumab) is a proposed biosimilar to Humira. AVT02 is produced at a 100 mg/mL concentration with a citrate-free formulation.
The aim of this study was to compare the efficacy, safety and immunogenicity of AVT02 versus Humira in subjects with moderate to severe chronic plaque psoriasis.
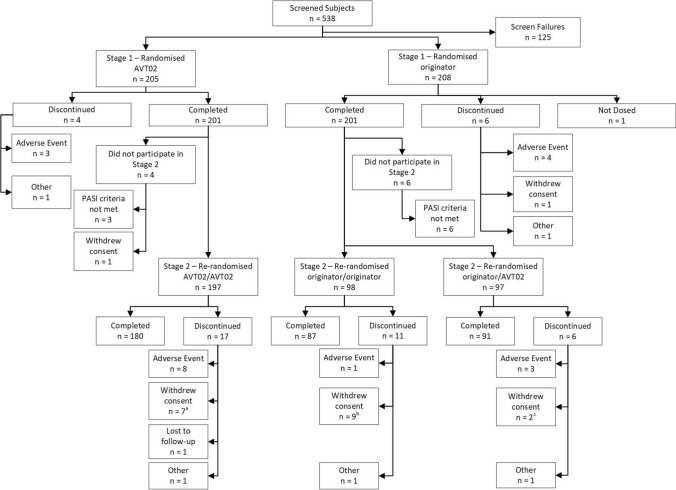
This double-blind, randomised, parallel group, active control study of adult subjects compared (at a 1:1 ratio) AVT02 with originator adalimumab 80 mg subcutaneously in Week 1, then 40 mg every other week. At Week 16, subjects who had received originator adalimumab were re-randomised at a 1:1 ratio to continue receiving originator adalimumab, or to switch to AVT02, every other week until Week 48, with final efficacy endpoint at Week 50. Subjects who initially received AVT02 continued to receive AVT02 from Week 16 to Week 48. The primary endpoint was percentage improvement in Psoriasis Area and Severity Index (PASI) score at Week 16. Secondary efficacy endpoints included percentage improvement in PASI score at additional timepoints, change from baseline in Dermatology Life Quality Index (DLQI) score and number and percentage of subjects achieving static Physician's Global Assessment (sPGA) responses of 'clear' or 'almost clear'. Additional secondary endpoints included comparison of adverse event profiles, anti-drug antibodies and neutralising antibodies, and serum trough levels of adalimumab at steady state.
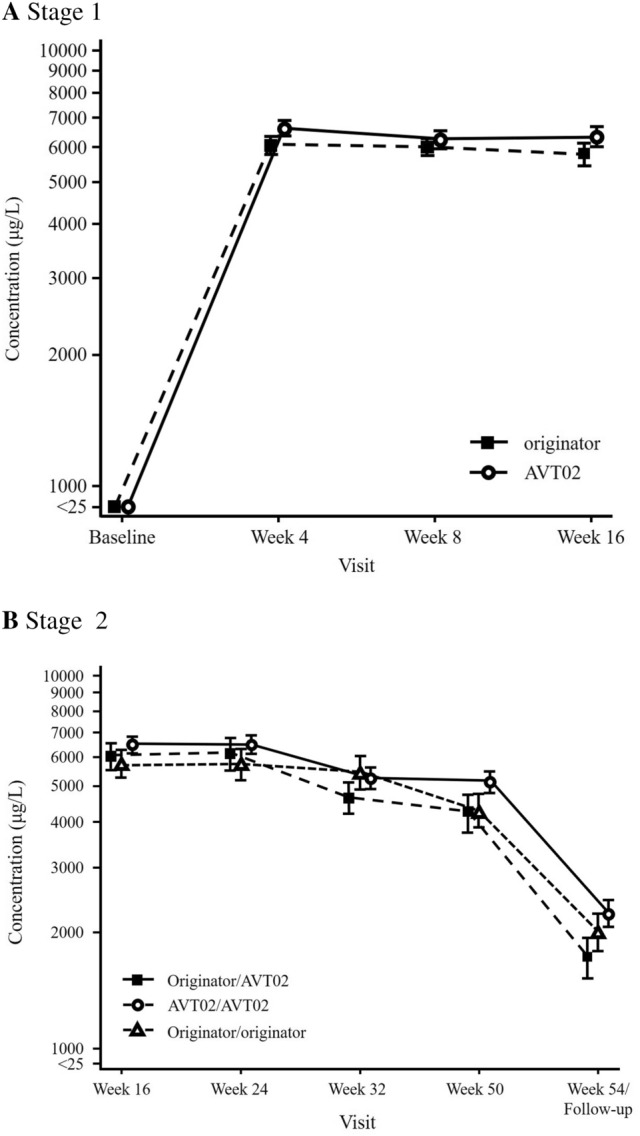
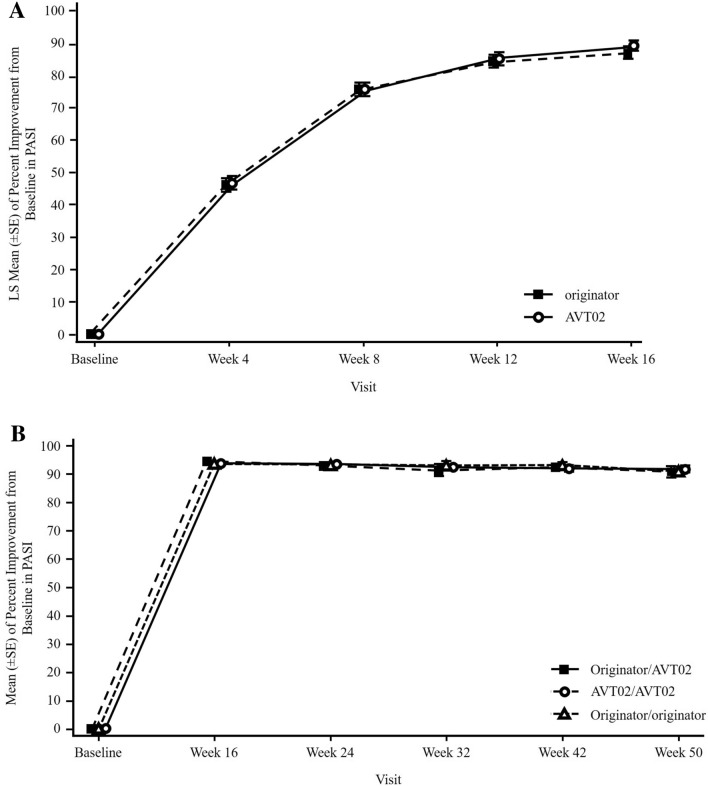
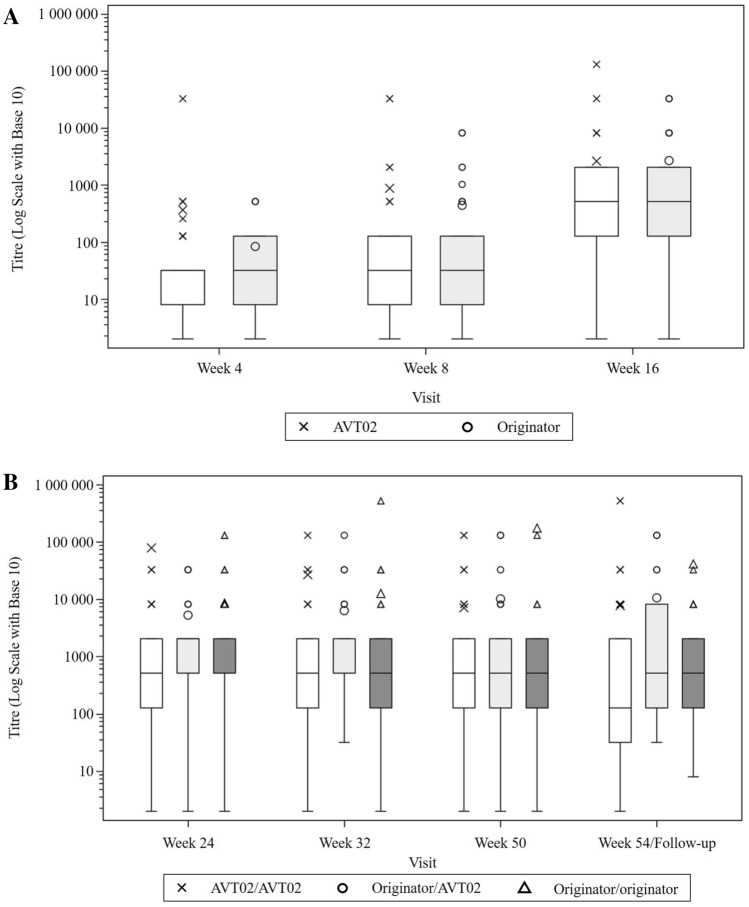
A total of 413 subjects were randomised (205 to AVT02 and 208 to originator). The percentage improvement in PASI score at Week 16 was 91.6% for AVT02-treated subjects and 89.6% for originator adalimumab. The 90% confidence intervals for the primary endpoint were within the pre-defined equivalence margin of ±10% (90% CI - 0.76 to 5.29; 95% CI - 1.34 to 5.88), and a comparable pattern for DLQI score (11.4-point and 10.6-point improvement in AVT02-treated and originator adalimumab-treated groups, respectively) and sPGA (90.5% in both groups achieving 'clear' or 'almost clear') at Week 16 supported the assessment. Efficacy persisted through Week 50 of the study in all treatment groups, including those who switched from originator adalimumab to AVT02, for percent improvement in PASI score, quality-of-life assessment and sPGA. The safety, tolerability and immunogenicity profiles between AVT02 and originator adalimumab were similar at Week 16, and this persisted in the switched and continued groups through Week 50.
Objective and subjective measures of efficacy supported the evaluation of biosimilarity between AVT02 and originator adalimumab at Week 16 and until Week 50, in switched and continued treatment groups. AVT02 was safe and well tolerated, with a safety and immunogenicity profile similar to that observed in originator adalimumab with no clinically meaningful difference between the two.
EudraCT: 2017-003367-35; ClinicalTrials.gov: NCT03849404.
AVT02(阿达木单抗)是一种拟议的与修美乐生物相似的药物。AVT02 以 100mg/ml 的浓度生产,采用无柠檬酸盐配方。
本研究旨在比较 AVT02 与 Humira 在中重度慢性斑块型银屑病患者中的疗效、安全性和免疫原性。
这是一项双盲、随机、平行组、活性对照研究,比较了成人受试者(1:1 比例)在第 1 周接受 AVT02 与阿达木单抗 80mg 皮下注射,然后每两周接受 40mg。在第 16 周,接受阿达木单抗的受试者按 1:1 的比例重新随机分组,继续接受阿达木单抗或每两周接受一次 AVT02,直到第 48 周,第 50 周为最终疗效终点。从第 16 周开始,最初接受 AVT02 的受试者继续接受 AVT02 至第 48 周。主要终点是第 16 周时 PASI 评分的改善百分比。次要疗效终点包括在其他时间点 PASI 评分的改善百分比、从基线到皮肤病生活质量指数(DLQI)评分的变化,以及达到静态医生总体评估(sPGA)“清除”或“几乎清除”反应的受试者数量和百分比。其他次要终点包括不良事件谱、抗药物抗体和中和抗体以及稳态时阿达木单抗血清浓度的比较。
共有 413 名受试者随机分组(205 名接受 AVT02 治疗,208 名接受阿达木单抗治疗)。第 16 周时,AVT02 治疗组 PASI 评分改善率为 91.6%,阿达木单抗治疗组为 89.6%。主要终点的 90%置信区间在预先定义的 10%等效性边界内(90%CI-0.76 至 5.29;95%CI-1.34 至 5.88),并且在第 16 周时,DLQI 评分(AVT02 治疗组和阿达木单抗治疗组分别改善 11.4 点和 10.6 点)和 sPGA(两组均有 90.5%的患者达到“清除”或“几乎清除”)的相似模式支持了这一评估。在研究的第 50 周,所有治疗组的疗效均持续存在,包括从阿达木单抗转换为 AVT02 的患者,PASI 评分、生活质量评估和 sPGA 的改善百分比。第 16 周时,AVT02 和阿达木单抗的安全性、耐受性和免疫原性特征相似,在第 50 周时,转换组和继续治疗组也保持了这一特征。
第 16 周和第 50 周时,AVT02 和阿达木单抗之间的疗效的客观和主观测量结果支持生物相似性评估,在转换组和继续治疗组中均有效。AVT02 安全且耐受良好,安全性和免疫原性特征与阿达木单抗相似,两者之间无临床意义的差异。
EudraCT:2017-003367-35;ClinicalTrials.gov:NCT03849404。