Catalano Claudio, Al Mughram Mohammed H, Guo Youzhong, Kellogg Glen E
Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, VA, USA.
Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA, USA.
Curr Res Struct Biol. 2021 Oct 1;3:239-256. doi: 10.1016/j.crstbi.2021.09.002. eCollection 2021.
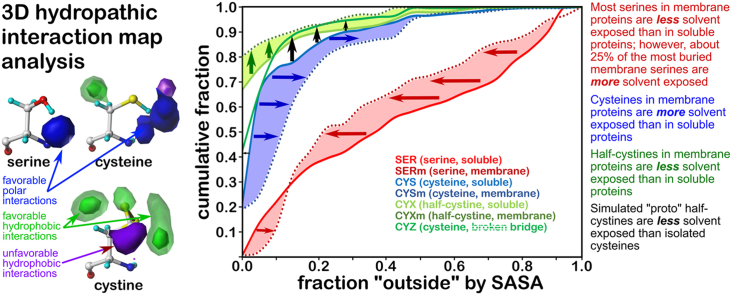
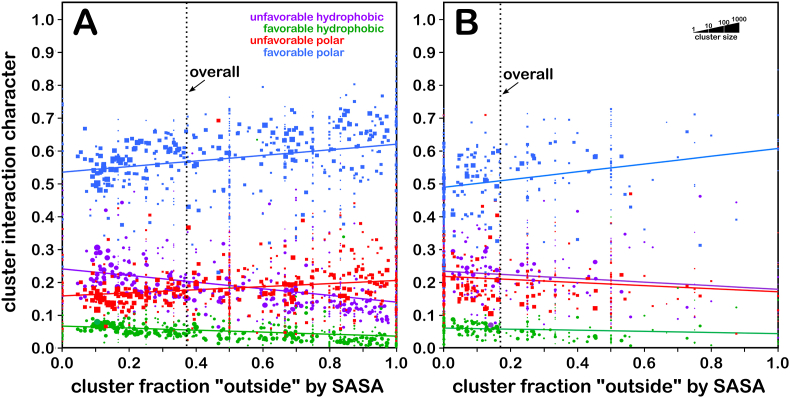
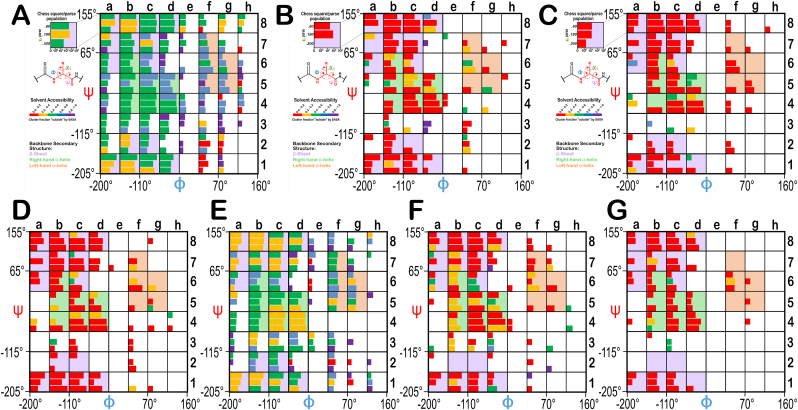
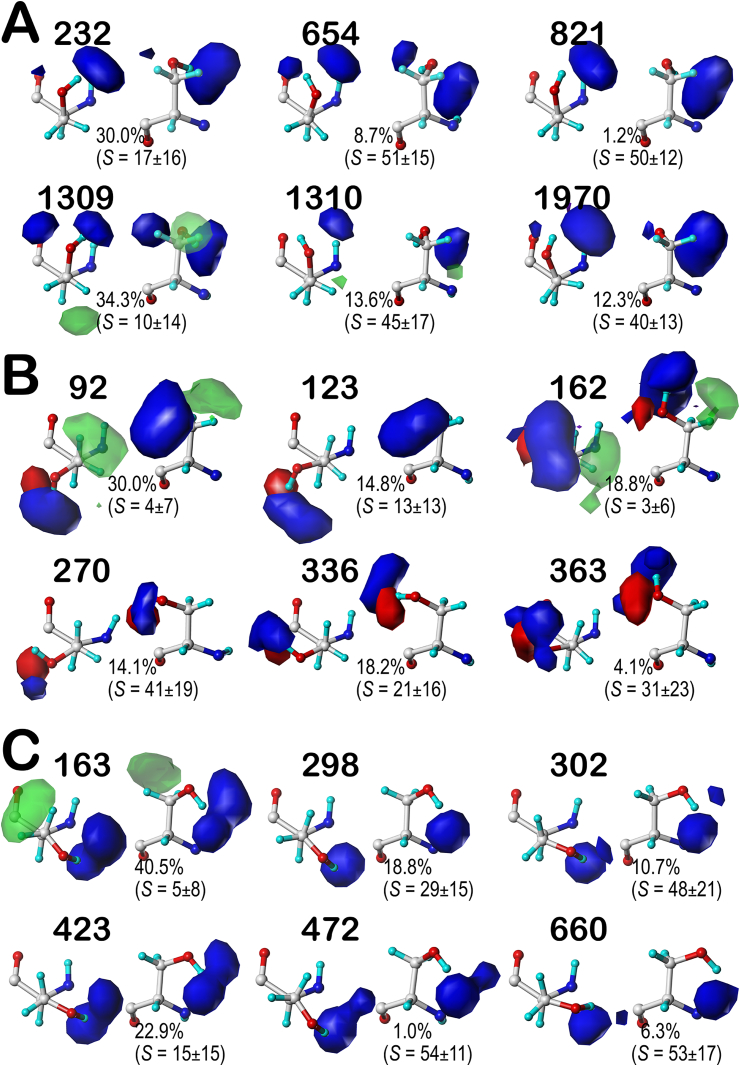
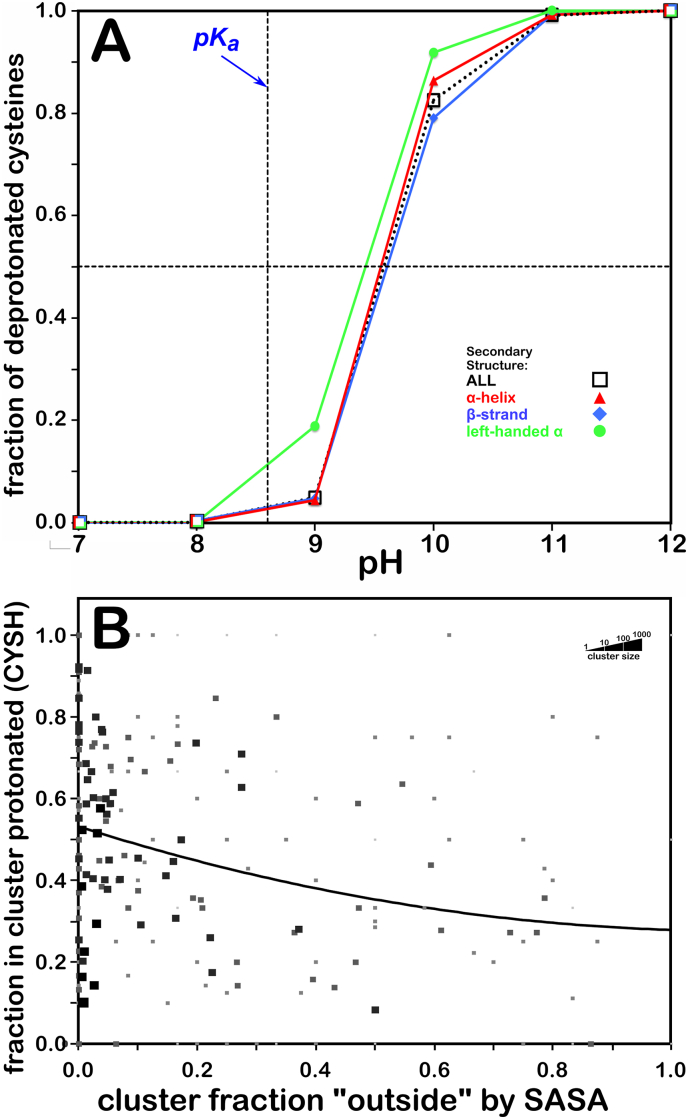
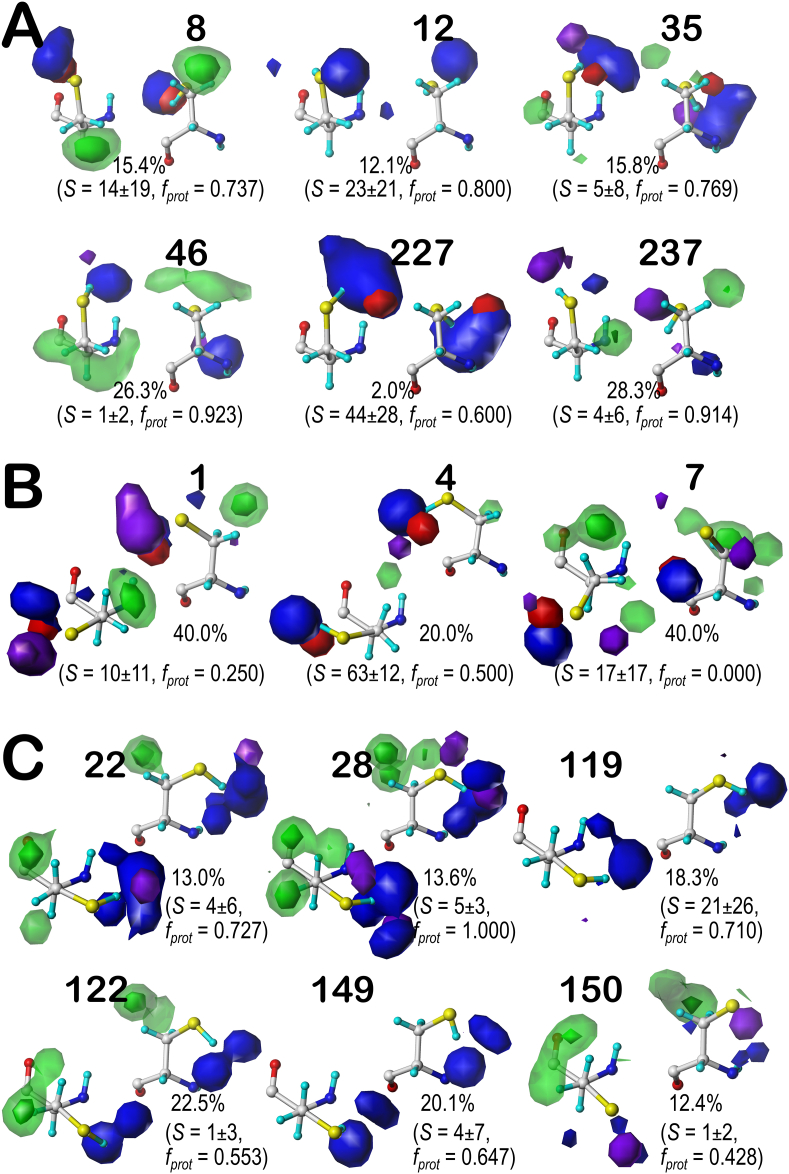
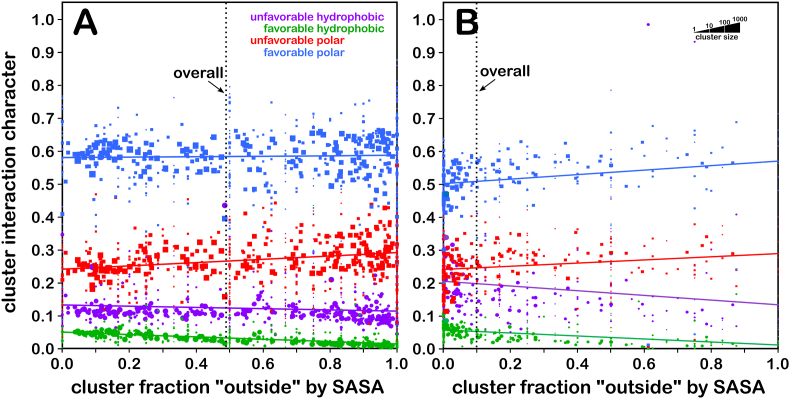
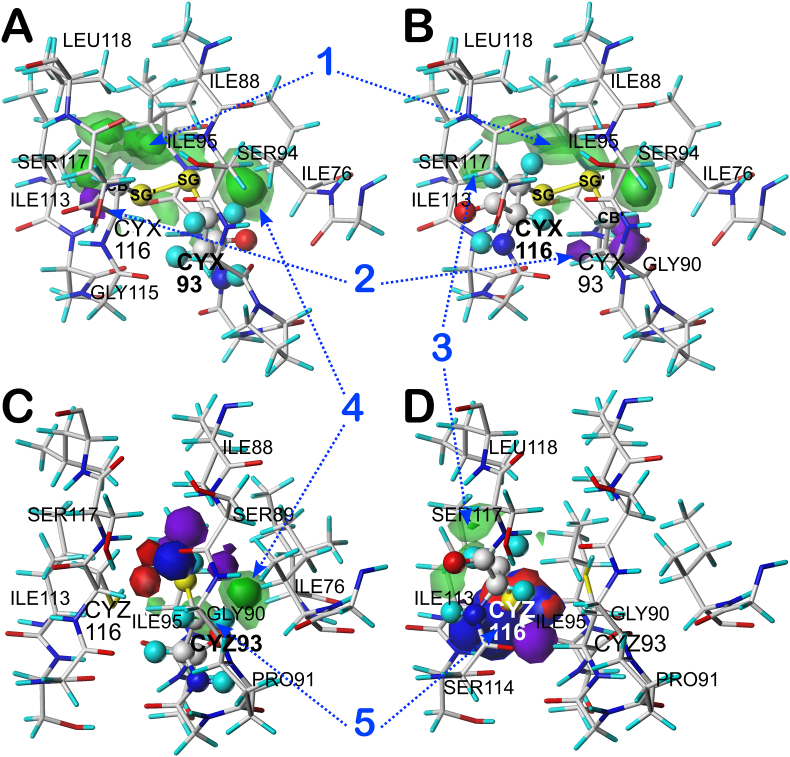
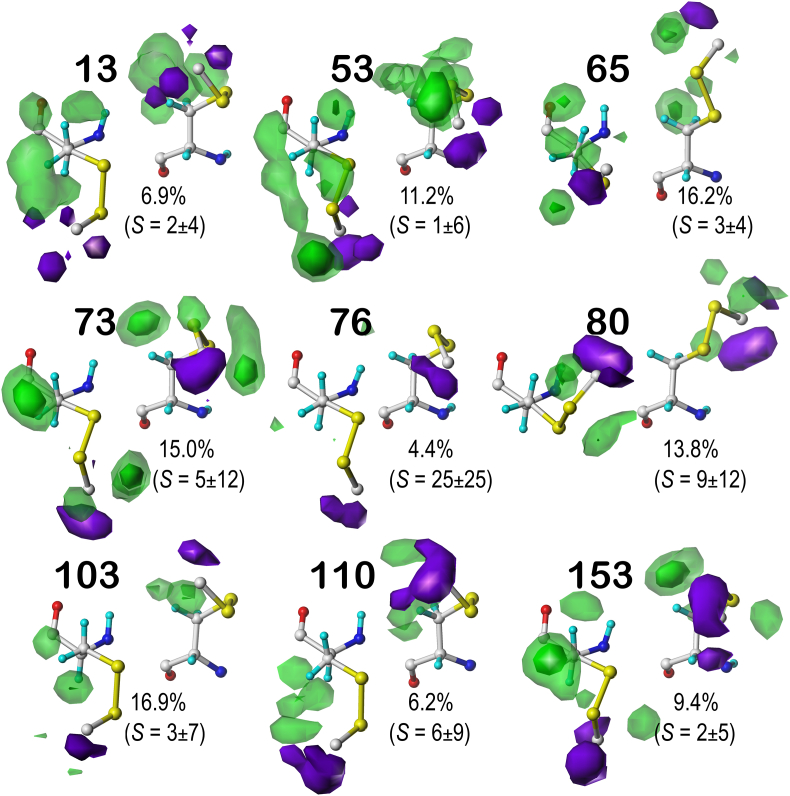
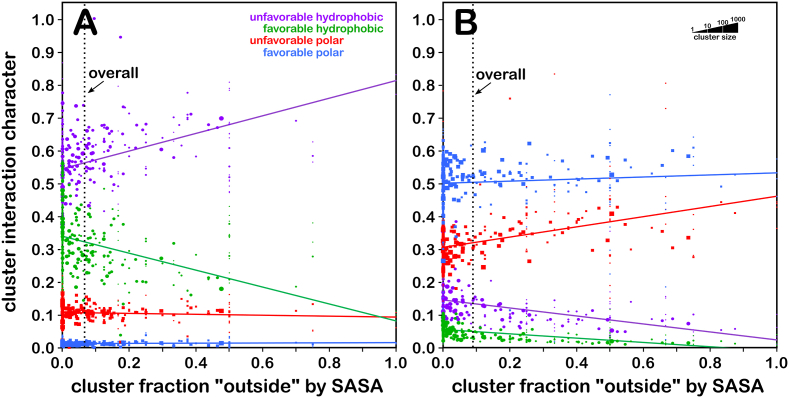
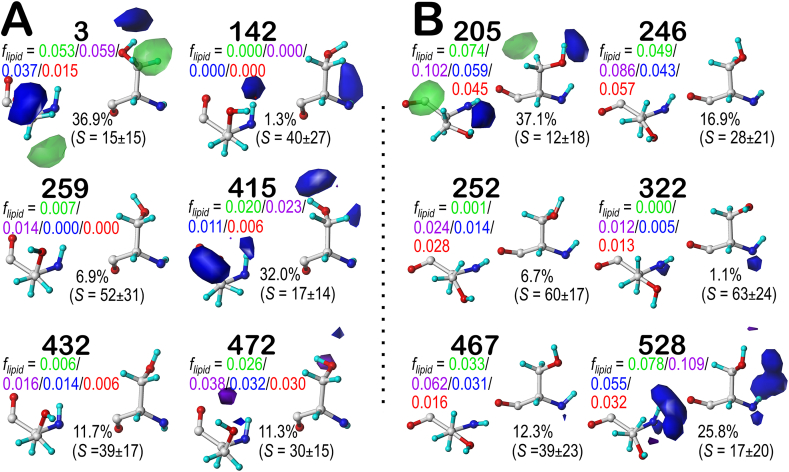
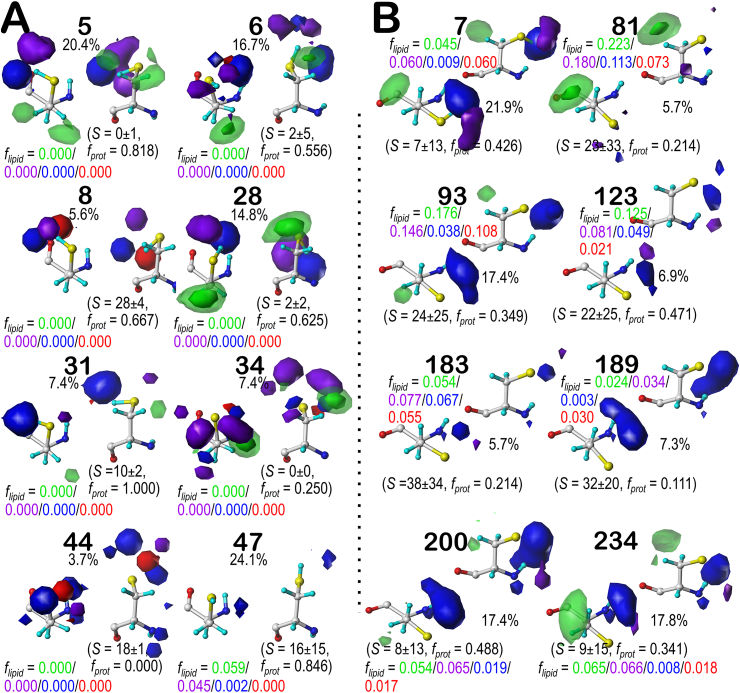
Atomic-resolution protein structural models are prerequisites for many downstream activities like structure-function studies or structure-based drug discovery. Unfortunately, this data is often unavailable for some of the most interesting and therapeutically important proteins. Thus, computational tools for building native-like structural models from less-than-ideal experimental data are needed. To this end, interaction homology exploits the character, strength and loci of the sets of interactions that define a structure. Each residue type has its own limited set of backbone angle-dependent interaction motifs, as defined by their environments. In this work, we characterize the interactions of serine, cysteine and S-bridged cysteine in terms of 3D hydropathic environment maps. As a result, we explore several intriguing questions. Are the environments different between the isosteric serine and cysteine residues? Do some environments promote the formation of cystine S-S bonds? With the increasing availability of structural data for water-insoluble membrane proteins, are there environmental differences for these residues between soluble and membrane proteins? The environments surrounding serine and cysteine residues are dramatically different: serine residues are about 50% solvent exposed, while cysteines are only 10% exposed; the latter are more involved in hydrophobic interactions although there are backbone angle-dependent differences. Our analysis suggests that one driving force for -S-S- bond formation is a rather substantial increase in burial and hydrophobic interactions in cystines. Serine and cysteine become less and more, respectively, solvent-exposed in membrane proteins. 3D hydropathic environment maps are an evolving structure analysis tool showing promise as elements in a new protein structure prediction paradigm.
原子分辨率的蛋白质结构模型是许多下游活动(如结构-功能研究或基于结构的药物发现)的先决条件。不幸的是,对于一些最有趣且在治疗上很重要的蛋白质,这些数据往往无法获得。因此,需要用于从不太理想的实验数据构建类似天然结构模型的计算工具。为此,相互作用同源性利用了定义结构的相互作用集的特征、强度和位点。每种残基类型都有其自身有限的一组依赖于主链角度的相互作用基序,这由它们的环境所定义。在这项工作中,我们根据三维疏水性质环境图来描述丝氨酸、半胱氨酸和S-桥连半胱氨酸的相互作用。结果,我们探讨了几个有趣的问题。等排的丝氨酸和半胱氨酸残基之间的环境是否不同?某些环境是否促进胱氨酸S-S键的形成?随着水不溶性膜蛋白结构数据的日益增多,这些残基在可溶性蛋白和膜蛋白之间是否存在环境差异?丝氨酸和半胱氨酸残基周围的环境有显著差异:丝氨酸残基约50%暴露于溶剂中,而半胱氨酸仅10%暴露;尽管存在主链角度依赖性差异,但后者更多地参与疏水相互作用。我们的分析表明,-S-S-键形成的一个驱动力是胱氨酸中埋藏和疏水相互作用的相当大增加。在膜蛋白中,丝氨酸和半胱氨酸分别变得更少和更多地暴露于溶剂中。三维疏水性质环境图是一种不断发展的结构分析工具,有望成为新的蛋白质结构预测范式中的元素。