Department of Clinical Laboratory, Peking University People's Hospital, Beijing, 100044, China.
UCL Genetics Institute, University College London, Gower Street, London, WC1E 6BT, UK.
Genome Med. 2021 Oct 28;13(1):171. doi: 10.1186/s13073-021-00992-x.
Methicillin-resistant Staphylococcus aureus (MRSA) is a major nosocomial pathogen subdivided into lineages termed sequence types (STs). Since the 1950s, successive waves of STs have appeared and replaced previously dominant lineages. One such event has been occurring in China since 2013, with community-associated (CA-MRSA) strains including ST59 largely replacing the previously dominant healthcare-associated (HA-MRSA) ST239. We previously showed that ST59 isolates tend to have a competitive advantage in growth experiments against ST239. However, the underlying genomic and phenotypic drivers of this replacement event are unclear.
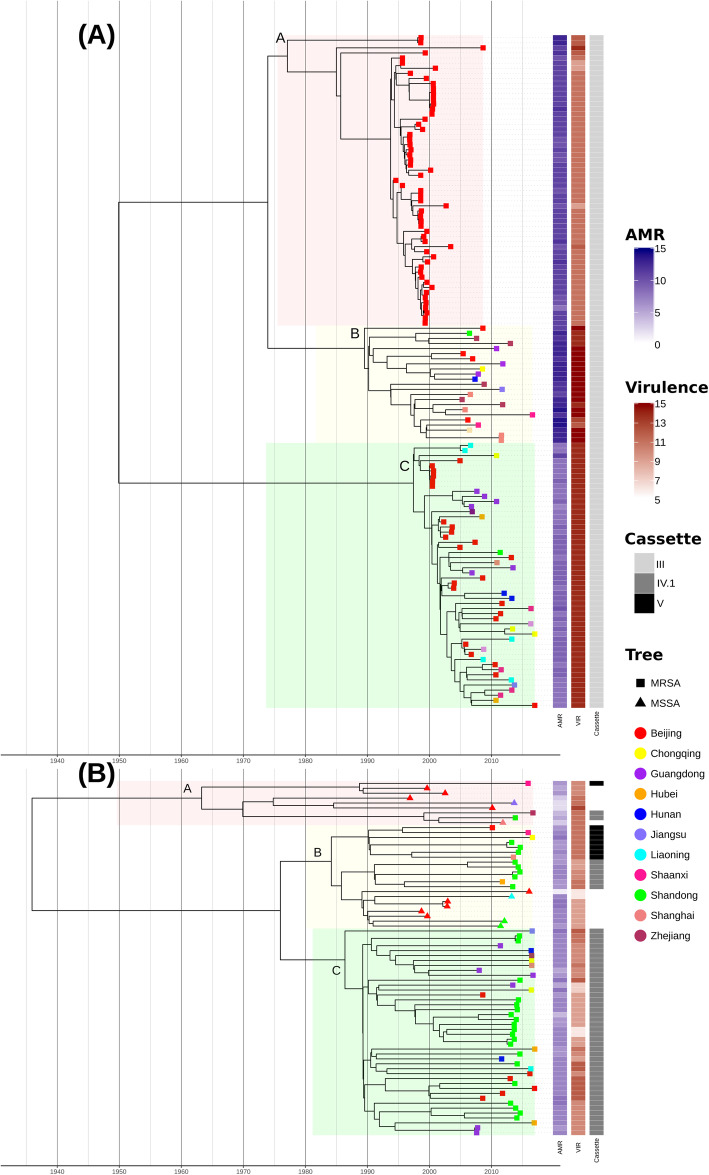
Here, we investigated the replacement of ST239 using whole-genome sequencing data from 204 ST239 and ST59 isolates collected in Chinese hospitals between 1994 and 2016. We reconstructed the evolutionary history of each ST and considered two non-mutually exclusive hypotheses for ST59 replacing ST239: antimicrobial resistance (AMR) profile and/or ability to colonise and persist in the environment through biofilm formation. We also investigated the differences in cytolytic activity, linked to higher virulence, between STs. We performed an association study using the presence and absence of accessory virulence genes.
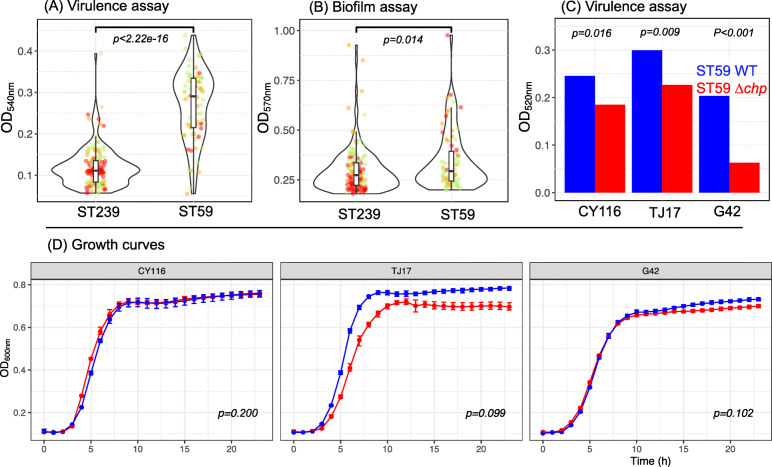
ST59 isolates carried fewer AMR genes than ST239 and showed no evidence of evolving towards higher AMR. Biofilm production was marginally higher in ST59 overall, though this effect was not consistent across sub-lineages so is unlikely to be a sole driver of replacement. Consistent with previous observations of higher virulence in CA-MRSA STs, we observed that ST59 isolates exhibit significantly higher cytolytic activity than ST239 isolates, despite carrying on average fewer putative virulence genes. Our association study identified the chemotaxis inhibitory protein (chp) as a strong candidate for involvement in the increased virulence potential of ST59. We experimentally validated the role of chp in increasing the virulence potential of ST59 by creating Δchp knockout mutants, confirming that ST59 can carry chp without a measurable impact on fitness.
Our results suggest that the ongoing replacement of ST239 by ST59 in China is not associated to higher AMR carriage or biofilm production. However, the increase in ST59 prevalence is concerning since it is linked to a higher potential for virulence, aided by the carriage of the chp gene.
耐甲氧西林金黄色葡萄球菌(MRSA)是一种主要的医院病原体,可分为称为序列类型(ST)的谱系。自 20 世纪 50 年代以来,相继出现了一波又一波的 ST,取代了先前占主导地位的谱系。自 2013 年以来,中国发生了这样的事件,包括社区相关(CA-MRSA)菌株 ST59 在内的菌株在很大程度上取代了先前占主导地位的医疗保健相关(HA-MRSA)ST239。我们之前曾表明,ST59 分离株在与 ST239 的生长实验中具有竞争优势。然而,这种替代事件的潜在基因组和表型驱动因素尚不清楚。
在这里,我们使用从 1994 年至 2016 年在中国医院收集的 204 株 ST239 和 ST59 分离株的全基因组测序数据研究了 ST239 的替代情况。我们重建了每个 ST 的进化历史,并考虑了 ST59 取代 ST239 的两个非相互排斥的假设:抗生素耐药性(AMR)谱和/或通过生物膜形成在环境中定植和持续存在的能力。我们还研究了与更高毒力相关的细胞溶解活性之间的 ST 差异。我们使用辅助毒力基因的存在和不存在进行了关联研究。
ST59 分离株携带的 AMR 基因少于 ST239,并且没有证据表明它们朝着更高 AMR 进化。总体而言,ST59 的生物膜产生略有增加,但这种效应在亚谱系之间并不一致,因此不太可能是替代的唯一驱动因素。与先前观察到 CA-MRSA ST 更高毒力的情况一致,我们观察到 ST59 分离株的细胞溶解活性明显高于 ST239 分离株,尽管平均携带的潜在毒力基因较少。我们的关联研究确定趋化抑制蛋白(chp)是参与 ST59 增加毒力潜力的候选基因。我们通过创建Δchp 敲除突变体实验验证了 chp 在增加 ST59 毒力潜力中的作用,证实 ST59 可以携带 chp,而不会对适应性产生可衡量的影响。
我们的结果表明,中国 ST239 被 ST59 取代与更高的 AMR 携带或生物膜产生无关。然而,由于携带 chp 基因,ST59 的流行率增加令人担忧,因为它与更高的毒力潜力有关。