BGI-Shenzhen, Shenzhen 518083, China.
China National GeneBank, BGI-Shenzhen, Shenzhen 518120, China.
Genome Res. 2019 May;29(5):798-808. doi: 10.1101/gr.245126.118. Epub 2019 Apr 2.
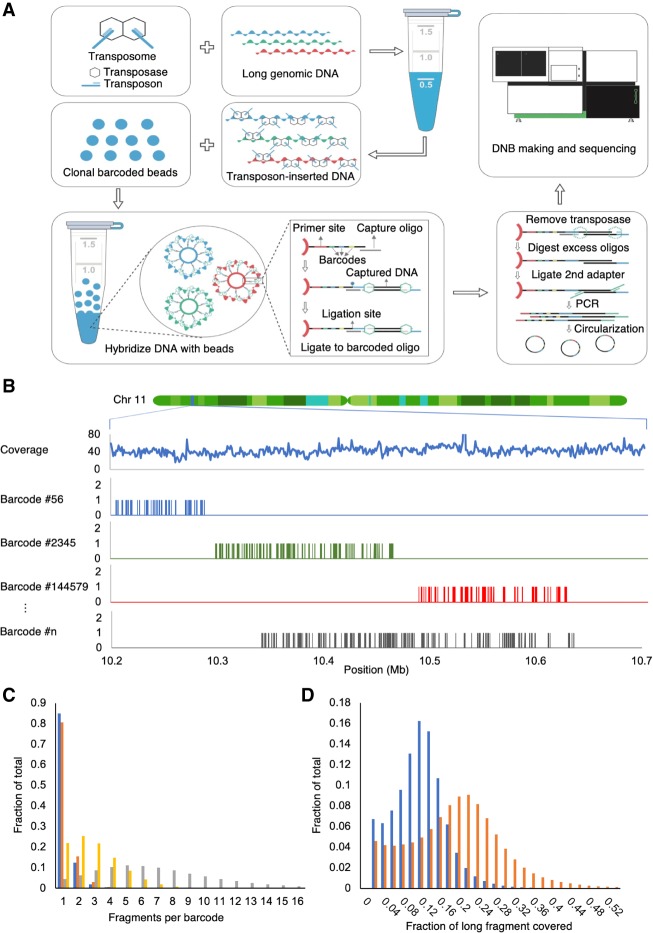
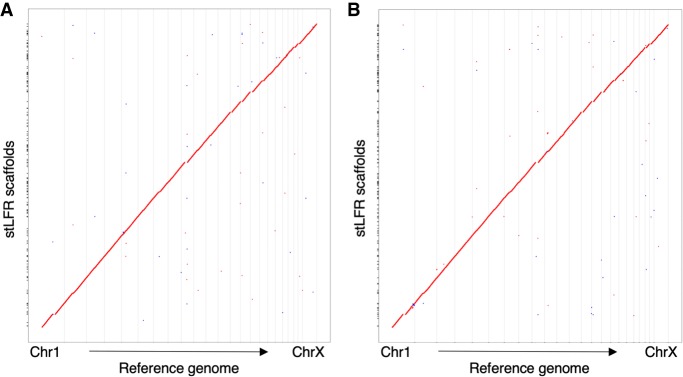
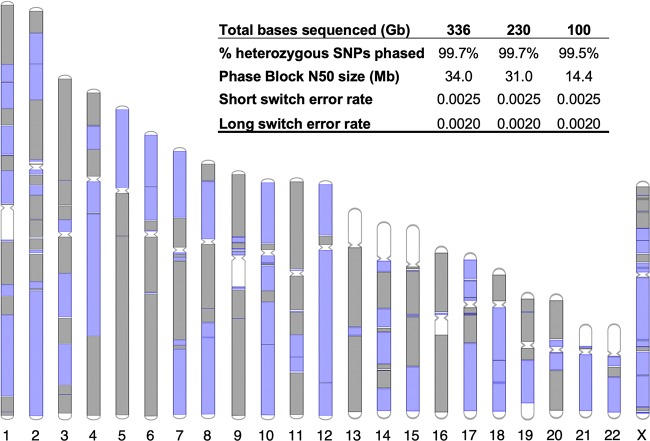
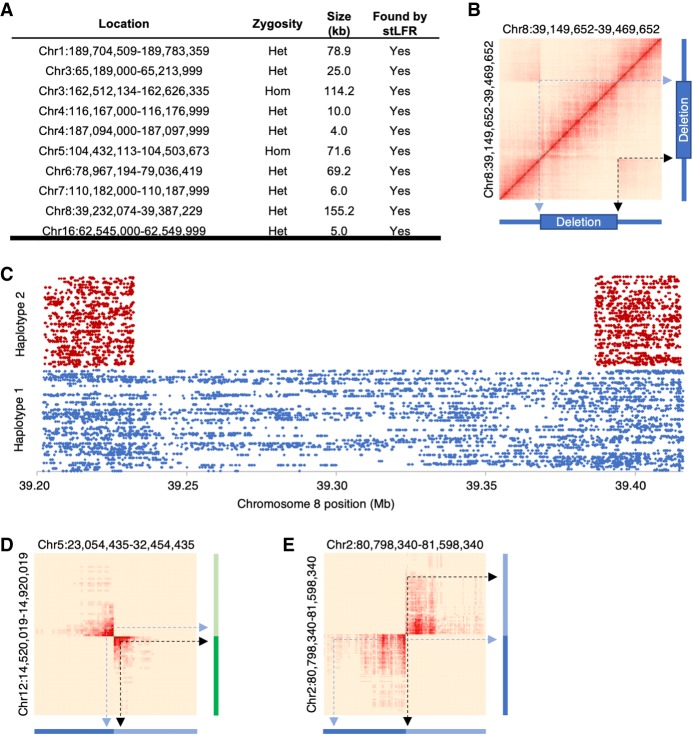
Here, we describe single-tube long fragment read (stLFR), a technology that enables sequencing of data from long DNA molecules using economical second-generation sequencing technology. It is based on adding the same barcode sequence to subfragments of the original long DNA molecule (DNA cobarcoding). To achieve this efficiently, stLFR uses the surface of microbeads to create millions of miniaturized barcoding reactions in a single tube. Using a combinatorial process, up to 3.6 billion unique barcode sequences were generated on beads, enabling practically nonredundant cobarcoding with 50 million barcodes per sample. Using stLFR, we demonstrate efficient unique cobarcoding of more than 8 million 20- to 300-kb genomic DNA fragments. Analysis of the human genome NA12878 with stLFR demonstrated high-quality variant calling and phase block lengths up to N50 34 Mb. We also demonstrate detection of complex structural variants and complete diploid de novo assembly of NA12878. These analyses were all performed using single stLFR libraries, and their construction did not significantly add to the time or cost of whole-genome sequencing (WGS) library preparation. stLFR represents an easily automatable solution that enables high-quality sequencing, phasing, SV detection, scaffolding, cost-effective diploid de novo genome assembly, and other long DNA sequencing applications.
在这里,我们描述了单管长片段读取(stLFR)技术,该技术可使用经济的第二代测序技术对长 DNA 分子的数据进行测序。它基于向原始长 DNA 分子的亚片段添加相同的条形码序列(DNA 共条形码)。为了有效地实现这一点,stLFR 使用微珠的表面在单个管中创建数百万个微型条形码反应。通过组合过程,在珠上生成多达 36 亿个独特的条形码序列,从而实现每个样本实际上无冗余的共条形码化,每个样本有 5000 万个条形码。使用 stLFR,我们展示了超过 800 万个 20-300kb 基因组 DNA 片段的高效独特共条形码化。对人类基因组 NA12878 的 stLFR 分析表明,高质量的变体调用和相位块长度高达 N50 34Mb。我们还展示了复杂结构变体的检测和 NA12878 的完整二倍体从头组装。所有这些分析都是使用单个 stLFR 文库进行的,其构建并没有显著增加全基因组测序(WGS)文库制备的时间或成本。stLFR 代表了一种易于自动化的解决方案,可实现高质量的测序、定相、SV 检测、支架、具有成本效益的二倍体从头基因组组装和其他长 DNA 测序应用。