Liu Xiao-Rong, Xu Xing-Xing, Lin Si-Mei, Fan Cui-Ying, Ye Ting-Ting, Tang Bin, Shi Yi-Wu, Su Tao, Li Bing-Mei, Yi Yong-Hong, Luo Jian-Hong, Liao Wei-Ping
Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, Institute of Neuroscience and Department of Neurology of the Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China.
Department of Physiology, Wenzhou Medical University, Wenzhou, China.
Front Mol Neurosci. 2021 Oct 14;14:720984. doi: 10.3389/fnmol.2021.720984. eCollection 2021.
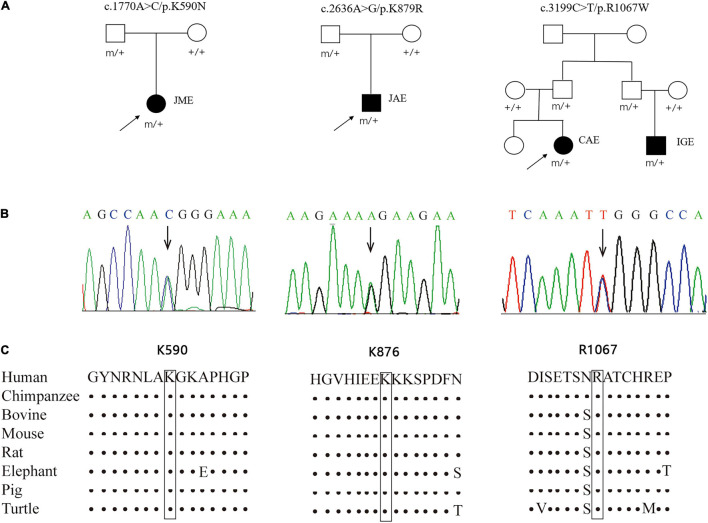
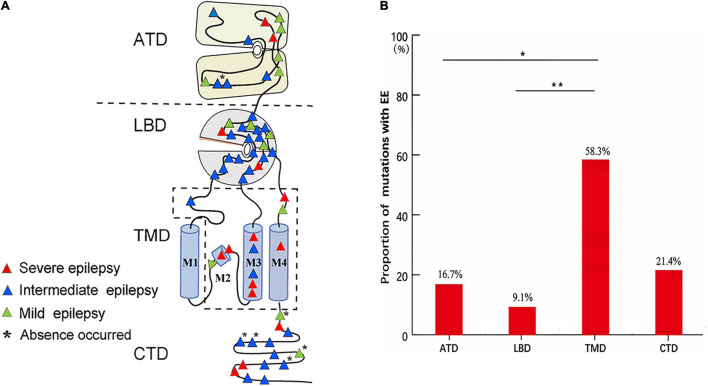
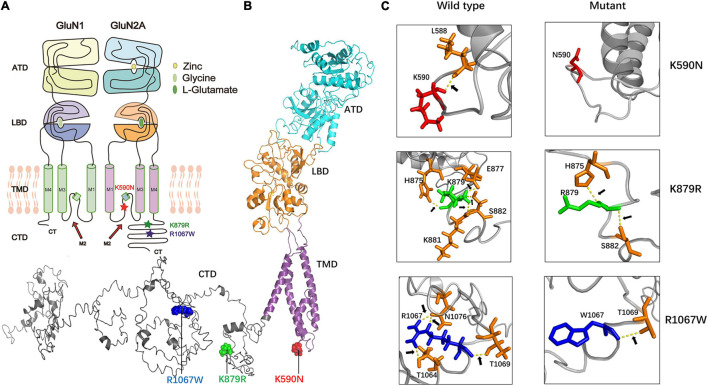
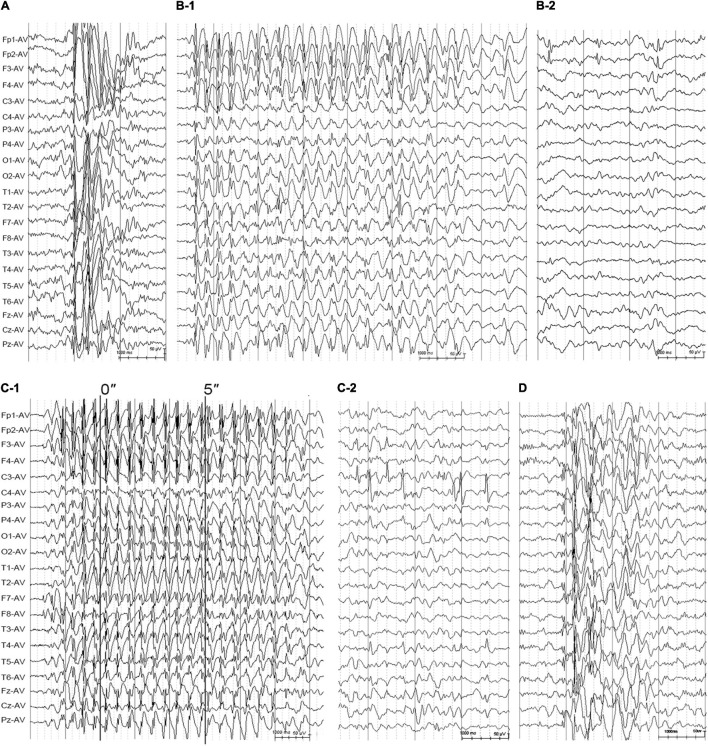
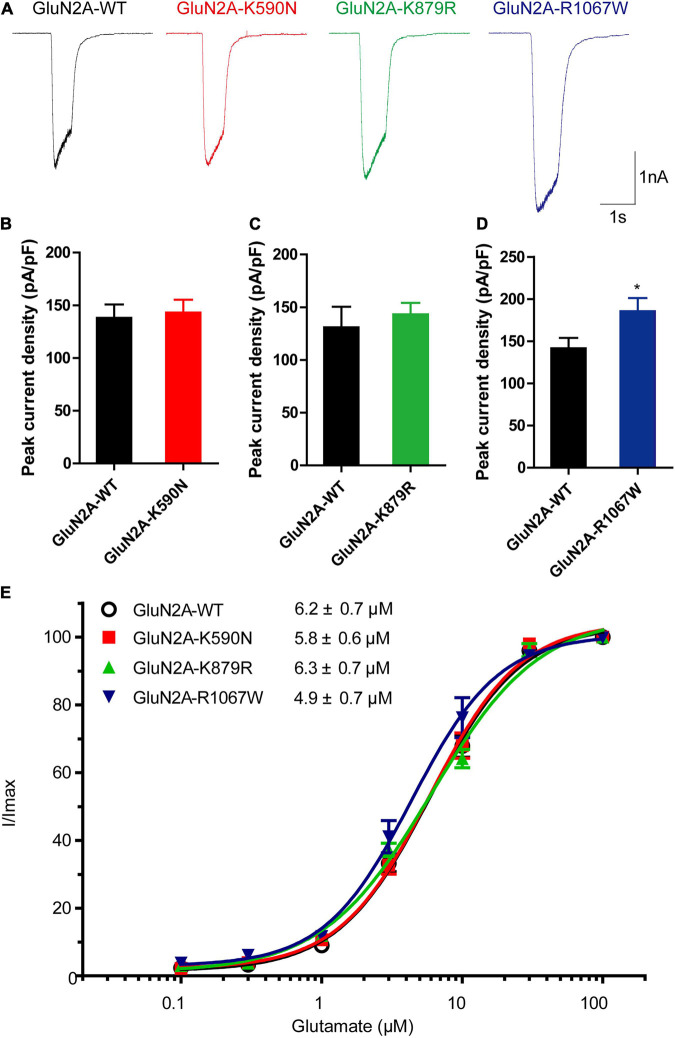
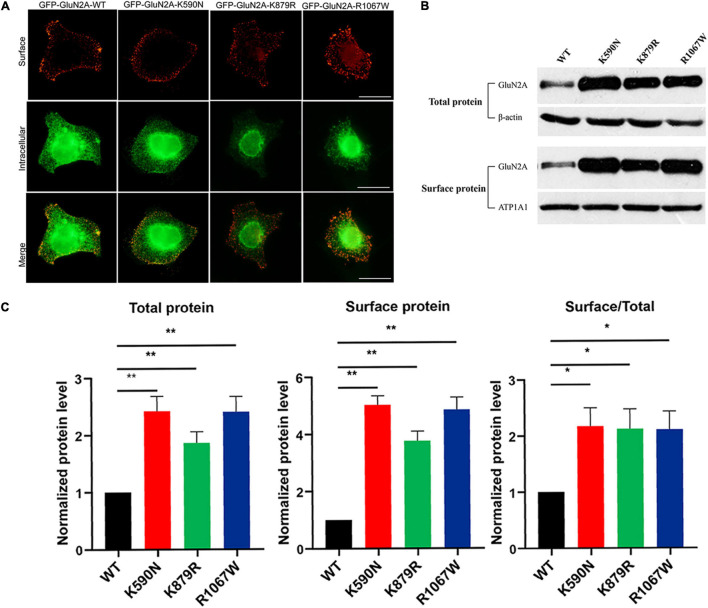
The objective of this study is to explore the role of gene in idiopathic generalized epilepsies and the potential underlying mechanism for phenotypic variation. Whole-exome sequencing was performed in a cohort of 88 patients with idiopathic generalized epilepsies. Electro-physiological alterations of the recombinant -methyl-D-aspartate receptors (NMDARs) containing GluN2A mutants were examined using two-electrode voltage-clamp recordings. The alterations of protein expression were detected by immunofluorescence staining and biotinylation. Previous studies reported that epilepsy related missense mutations were reviewed. The correlation among phenotypes, functional alterations, and molecular locations was analyzed. Three novel heterozygous missense mutations (c.1770A > C/p.K590N, c.2636A > G/p.K879R, and c.3199C > T/p.R1067W) were identified in three unrelated cases. Electrophysiological analysis demonstrated R1067W significantly increased the current density of GluN1/GluN2A NMDARs. Immunofluorescence staining indicated GluN2A mutants had abundant distribution in the membrane and cytoplasm. Western blotting showed the ratios of surface and total expression of the three GluN2A-mutants were significantly increased comparing to the wild type. Further analysis on the reported missense mutations demonstrated that mutations with severe gain-of-function were associated with epileptic encephalopathy, while mutations with mild gain of function were associated with mild phenotypes, suggesting a quantitative correlation between gain-of-function and phenotypic severity. The mutations located around transmembrane domains were more frequently associated with severe phenotypes and absence seizure-related mutations were mostly located in carboxyl-terminal domain, suggesting molecular sub-regional effects. This study revealed gene was potentially a candidate pathogenic gene of idiopathic generalized epilepsies. The functional quantitative correlation and the molecular sub-regional implication of mutations helped in explaining the relatively mild clinical phenotypes and incomplete penetrance associated with variants.
本研究的目的是探讨基因在特发性全身性癫痫中的作用以及表型变异的潜在机制。对88例特发性全身性癫痫患者进行了全外显子测序。使用双电极电压钳记录检测含有GluN2A突变体的重组N-甲基-D-天冬氨酸受体(NMDARs)的电生理改变。通过免疫荧光染色和生物素化检测蛋白质表达的改变。回顾了先前报道的与癫痫相关的错义突变。分析了表型、功能改变和分子位置之间的相关性。在3例无关病例中鉴定出3个新的杂合错义突变(c.1770A>C/p.K590N、c.2636A>G/p.K879R和c.3199C>T/p.R1067W)。电生理分析表明,R1067W显著增加了GluN1/GluN2A NMDARs的电流密度。免疫荧光染色表明,GluN2A突变体在细胞膜和细胞质中分布丰富。蛋白质免疫印迹显示,与野生型相比,三种GluN2A突变体的表面表达与总表达之比显著增加。对已报道的错义突变的进一步分析表明,功能严重增强的突变与癫痫性脑病相关,而功能轻度增强的突变与轻度表型相关,提示功能增强与表型严重程度之间存在定量相关性。位于跨膜结构域周围的突变更常与严重表型相关,失神发作相关突变大多位于羧基末端结构域,提示分子亚区域效应。本研究表明,该基因可能是特发性全身性癫痫的候选致病基因。突变的功能定量相关性和分子亚区域意义有助于解释与该基因变异相关的相对较轻的临床表型和不完全外显率。