Department of Dermatology, Hunan Key Laboratory of Medical Epigenomics, The Second Xiangya Hospital of Central South University, Changsha, China.
Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing, China.
Front Immunol. 2021 Oct 15;12:691304. doi: 10.3389/fimmu.2021.691304. eCollection 2021.
Increasing evidence suggests that the gut microbiome plays a role in the pathogenesis of allergy and autoimmunity. The association between abnormalities in the gut microbiota and chronic spontaneous urticaria (CSU) remains largely undefined.
Fecal samples were obtained from 39 patients with CSU and 40 healthy controls (HCs). 16S ribosomal RNA (rRNA) gene sequencing (39 patients with CSU and 40 HCs) and untargeted metabolomics (12 patients with CSU and 12 HCs) were performed to analyze the compositional and metabolic alterations of the gut microbiome in CSU patients and HCs.
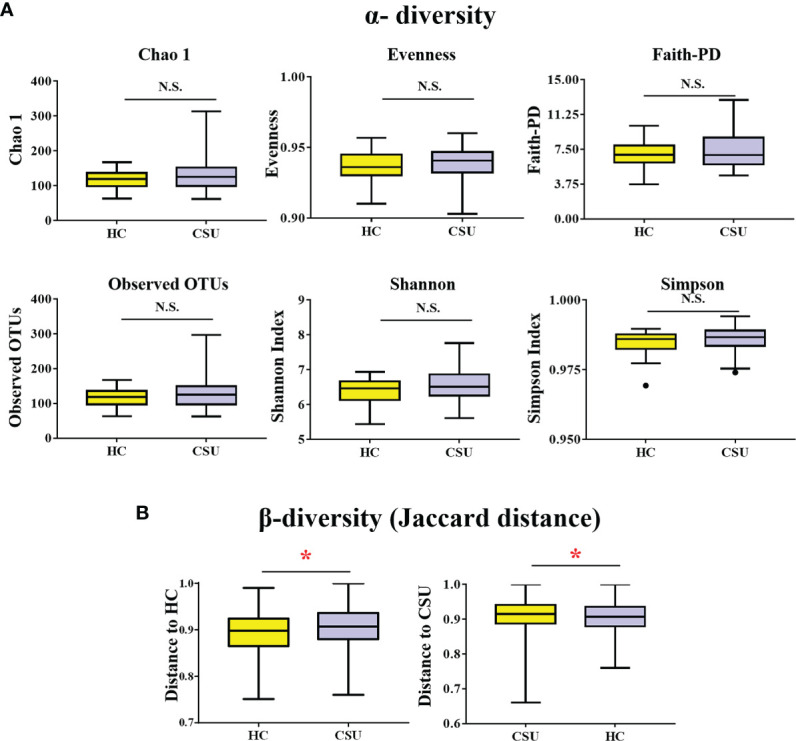
The 16S rRNA gene sequencing results showed a significant difference in the β-diversity of the gut microbiota, presented as the Jaccard distance, between CSU patients and HCs. No significant differences were found in the α-diversity of the gut microbiota between patients and HCs. At the phylum level, the major bacteria in the gut microbiome of patients with CSU were Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria. At the genus level, , , and were significantly increased and was decreased in patients with CSU. PICRUSt and correlation analysis indicated that , , and were positively related to G protein-coupled receptors. Metabolomic analysis showed that α-mangostin and glycyrrhizic acid were upregulated and that 3-indolepropionic acid, xanthine, and isobutyric acid were downregulated in patients with CSU. Correlation analysis between the intestinal microbiota and metabolites suggested that there was a positive correlation between and α-mangostin.
This study suggests that disturbances in the gut microbiome composition and metabolites and their crosstalk or interaction may participate in the pathogenesis of CSU.
越来越多的证据表明,肠道微生物群在过敏和自身免疫的发病机制中起作用。肠道微生物群的异常与慢性自发性荨麻疹(CSU)之间的关联在很大程度上仍未得到明确界定。
从 39 例 CSU 患者和 40 例健康对照者(HCs)中获得粪便样本。对 39 例 CSU 患者和 40 例 HCs 进行 16S 核糖体 RNA(rRNA)基因测序(39 例 CSU 患者和 40 例 HCs)和非靶向代谢组学(12 例 CSU 患者和 12 例 HCs),以分析 CSU 患者和 HCs 肠道微生物组的组成和代谢变化。
16S rRNA 基因测序结果显示,CSU 患者与 HCs 之间肠道微生物群的β多样性(以 Jaccard 距离表示)存在显著差异。患者与 HCs 之间肠道微生物群的α多样性无显著差异。在门水平上,CSU 患者肠道微生物群的主要细菌为厚壁菌门、拟杆菌门、变形菌门和放线菌门。在属水平上, 、 、 和 显著增加,而 减少。PICRUSt 和相关性分析表明, 、 、 和 G 蛋白偶联受体呈正相关。代谢组学分析显示,CSU 患者的 α-倒捻子素和甘草酸上调,而 3-吲哚丙酸、黄嘌呤和异丁酸下调。肠道微生物群和代谢物之间的相关性分析表明, 和 α-倒捻子素之间存在正相关。
本研究表明,肠道微生物群组成和代谢物的紊乱及其相互作用或相互作用可能参与 CSU 的发病机制。