Chen Shuan, Jung Yousung
Department of Chemical and Biomolecular Engineering (BK21 four), KAIST, 291 Daehak-ro, Daejeon 34141, South Korea.
JACS Au. 2021 Aug 5;1(10):1612-1620. doi: 10.1021/jacsau.1c00246. eCollection 2021 Oct 25.
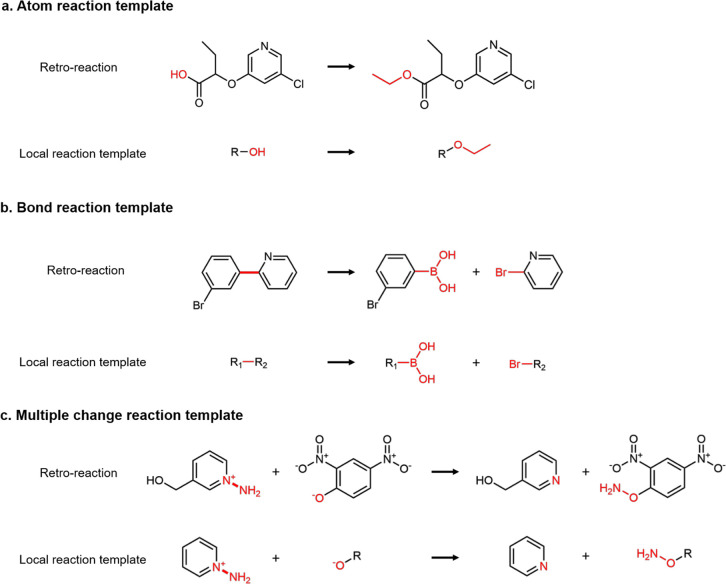
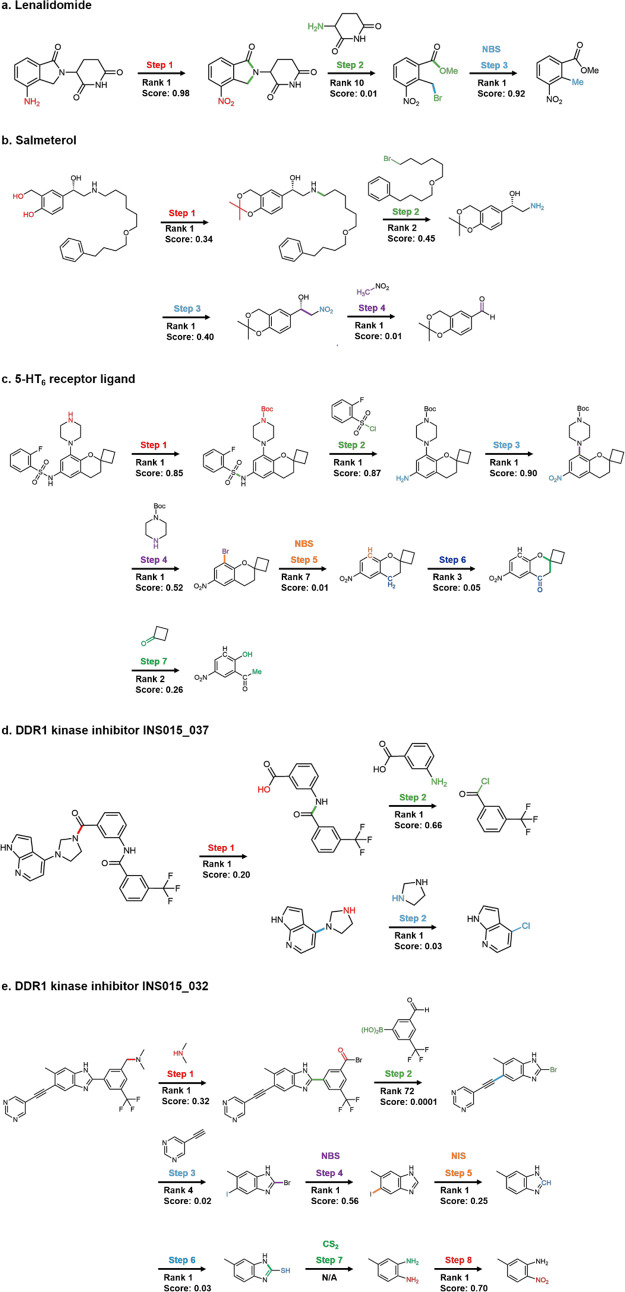
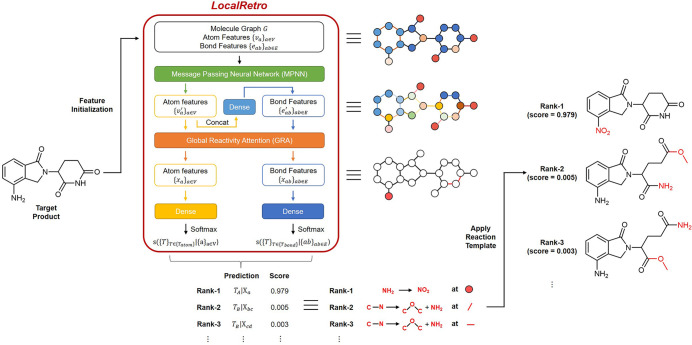
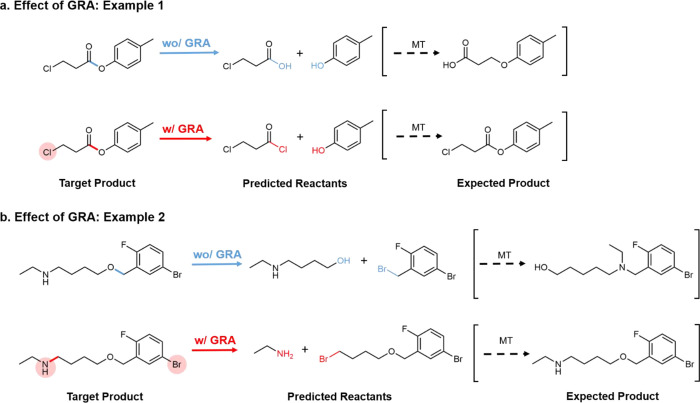
As a fundamental problem in chemistry, retrosynthesis aims at designing reaction pathways and intermediates for a target compound. The goal of artificial intelligence (AI)-aided retrosynthesis is to automate this process by learning from the previous chemical reactions to make new predictions. Although several models have demonstrated their potentials for automated retrosynthesis, there is still a significant need to further enhance the prediction accuracy to a more practical level. Here we propose a local retrosynthesis framework called , motivated by the chemical intuition that the molecular changes occur mostly locally during the chemical reactions. This differs from nearly all existing retrosynthesis methods that suggest reactants based on the global structures of the molecules, often containing fine details not directly relevant to the reactions. This local concept yields local reaction templates involving the atom and bond edits. Because the remote functional groups can also affect the overall reaction path as a secondary aspect, the proposed locally encoded retrosynthesis model is then further refined to account for the nonlocal effects of chemical reaction through a global attention mechanism. Our model shows a promising 89.5 and 99.2% round-trip accuracy at top-1 and top-5 predictions for the USPTO-50K dataset containing 50 016 reactions. We further demonstrate the validity of on a large dataset containing 479 035 reactions (UTPTO-MIT) with comparable round-trip top-1 and top-5 accuracy of 87.0 and 97.4%, respectively. The practical application of the model is also demonstrated by correctly predicting the synthesis pathways of five drug candidate molecules from various literature.
作为化学领域的一个基本问题,逆合成旨在为目标化合物设计反应路径和中间体。人工智能辅助逆合成的目标是通过学习以往的化学反应来自动完成这一过程,从而做出新的预测。尽管有几种模型已经展示了它们在自动逆合成方面的潜力,但仍有很大的需求将预测准确率进一步提高到更实用的水平。在此,我们提出了一种名为 的局部逆合成框架,其灵感来源于化学反应中分子变化大多发生在局部的化学直觉。这与几乎所有现有的逆合成方法不同,后者基于分子的整体结构来提出反应物,通常包含与反应不直接相关的精细细节。这种局部概念产生了涉及原子和键编辑的局部反应模板。由于远程官能团作为次要方面也会影响整体反应路径,因此通过全局注意力机制对所提出的局部编码逆合成模型进行进一步优化,以考虑化学反应的非局部效应。对于包含50016个反应的USPTO - 50K数据集,我们的模型在top - 1和top - 5预测时分别显示出有前景的89.5%和99.2%的往返准确率。我们还在包含479035个反应的大型数据集(UTPTO - MIT)上证明了 的有效性,其往返top - 1和top - 5准确率分别为87.0%和97.4%,具有可比性。通过正确预测来自各种文献的五个候选药物分子的合成路径,也证明了该模型的实际应用。