Division of Biology and Biological Engineering, California Institute of Technology, 1200 E. California Blvd, Pasadena, CA, 91125, USA.
Medically Associated Science and Technology (MAST) Program, Cedars-Sinai Medical Center, Los Angeles, CA, 90048, USA.
Microbiome. 2021 Nov 2;9(1):214. doi: 10.1186/s40168-021-01162-2.
Upper gastrointestinal (GI) disorders and abdominal pain afflict between 12 and 30% of the worldwide population and research suggests these conditions are linked to the gut microbiome. Although large-intestine microbiota have been linked to several GI diseases, the microbiota of the human small intestine and its relation to human disease has been understudied. The small intestine is the major site for immune surveillance in the gut, and compared with the large intestine, it has greater than 100 times the surface area and a thinner and more permeable mucus layer.
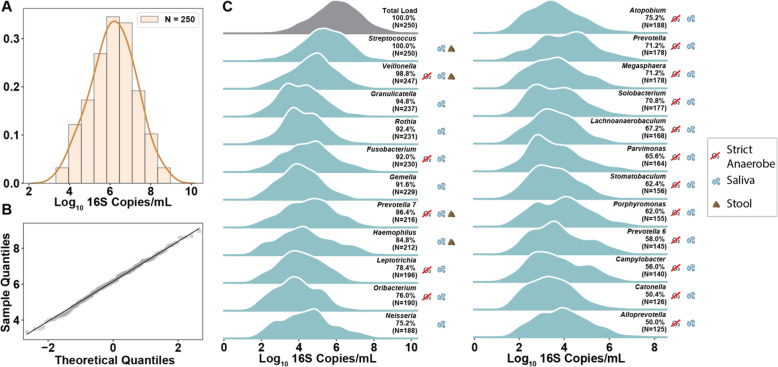
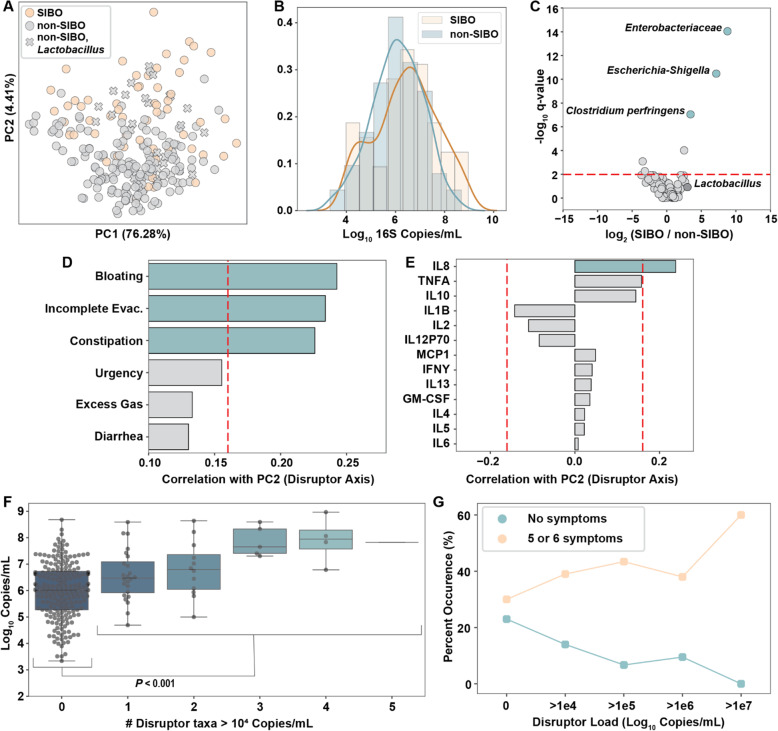
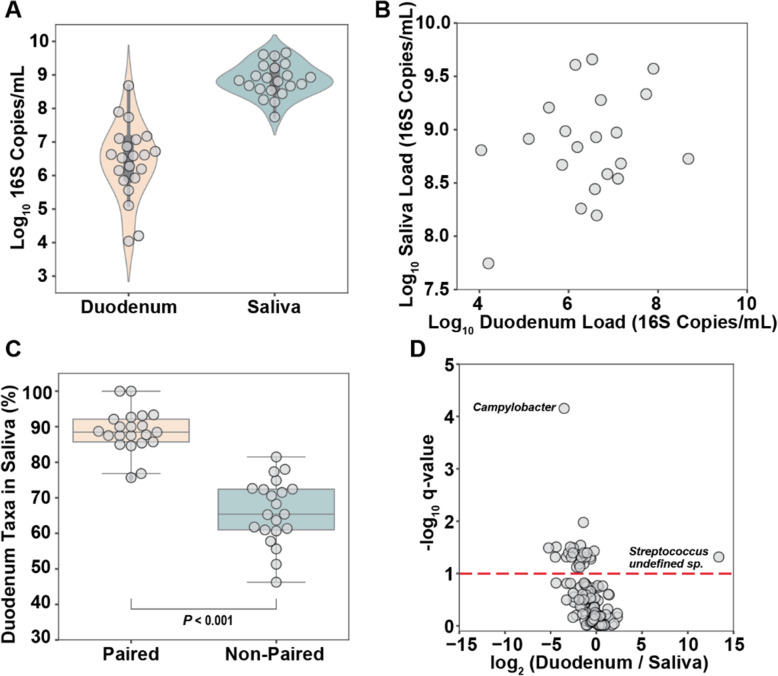
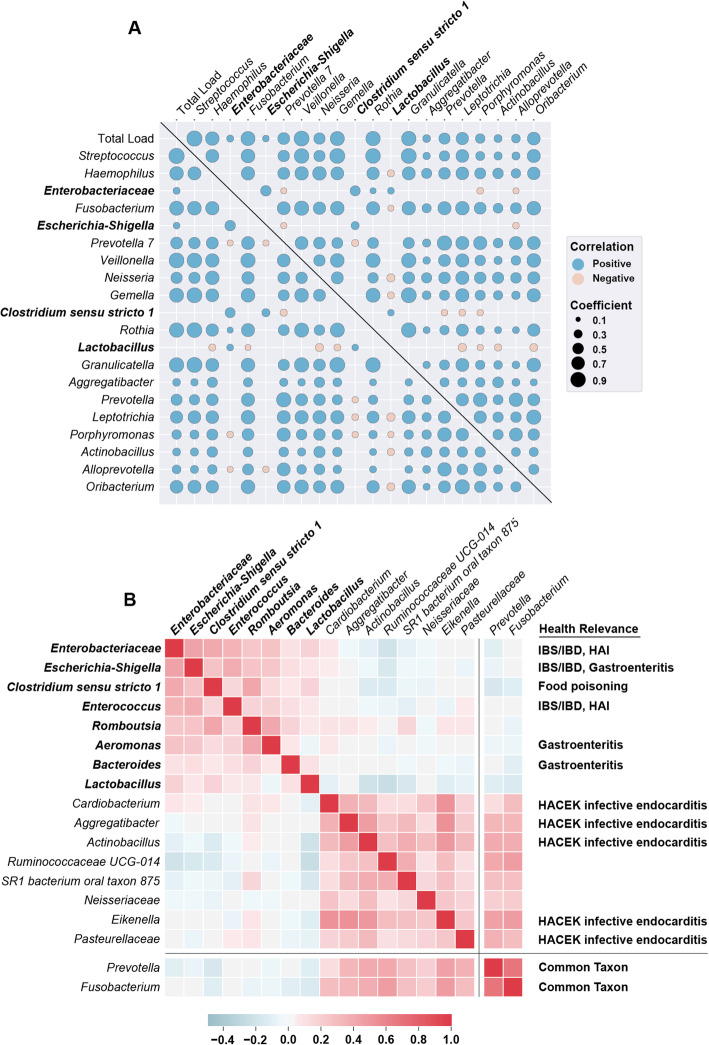
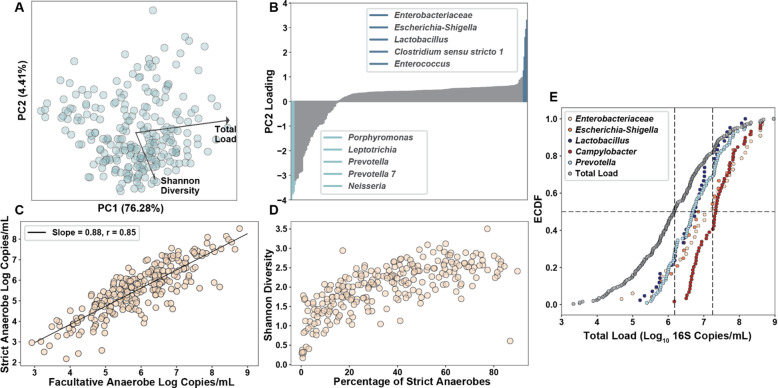
Using quantitative sequencing, we evaluated total and taxon-specific absolute microbial loads from 250 duodenal-aspirate samples and 21 paired duodenum-saliva samples from participants in the REIMAGINE study. Log-transformed total microbial loads spanned 5 logs and were normally distributed. Paired saliva-duodenum samples suggested potential transmission of oral microbes to the duodenum, including organisms from the HACEK group. Several taxa, including Klebsiella, Escherichia, Enterococcus, and Clostridium, seemed to displace strict anaerobes common in the duodenum, so we refer to these taxa as disruptors. Disruptor taxa were enriched in samples with high total microbial loads and in individuals with small intestinal bacterial overgrowth (SIBO). Absolute loads of disruptors were associated with more severe GI symptoms, highlighting the value of absolute taxon quantification when studying small-intestine health and function.
This study provides the largest dataset of the absolute abundance of microbiota from the human duodenum to date. The results reveal a clear relationship between the oral microbiota and the duodenal microbiota and suggest an association between the absolute abundance of disruptor taxa, SIBO, and the prevalence of severe GI symptoms. Video Abstract.
全球有 12%至 30%的人口患有上消化道(GI)疾病和腹痛,研究表明这些疾病与肠道微生物群有关。尽管大肠微生物群与几种 GI 疾病有关,但人类小肠的微生物群及其与人类疾病的关系尚未得到充分研究。小肠是肠道中免疫监测的主要部位,与大肠相比,小肠的表面积大 100 多倍,黏液层更薄且更具渗透性。
我们使用定量测序评估了来自 REIMAGINE 研究的 250 份十二指肠抽吸样本和 21 对十二指肠-唾液样本的总微生物负荷和分类群特异性绝对微生物负荷。对数转换后的总微生物负荷跨越 5 个对数,呈正态分布。唾液-十二指肠配对样本提示口腔微生物可能传播到十二指肠,包括 HACEK 组的微生物。包括克雷伯氏菌、大肠杆菌、肠球菌和梭菌在内的几种分类群似乎取代了常见于十二指肠的严格厌氧菌,因此我们将这些分类群称为“破坏者”。破坏者分类群在总微生物负荷较高的样本和小肠细菌过度生长(SIBO)个体中更为丰富。破坏者的绝对负荷与更严重的 GI 症状相关,这突出了在研究小肠健康和功能时绝对分类群定量的价值。
本研究提供了迄今为止最大的人类十二指肠微生物群绝对丰度数据集。结果显示口腔微生物群和十二指肠微生物群之间存在明显关系,并提示破坏者分类群的绝对丰度、SIBO 与严重 GI 症状的患病率之间存在关联。