Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA, USA.
Division of Chemistry and Chemical Engineering, California Institute of Technology, 1200 E. California Blvd, Pasadena, CA, USA.
Microbiome. 2020 Feb 12;8(1):19. doi: 10.1186/s40168-020-0785-4.
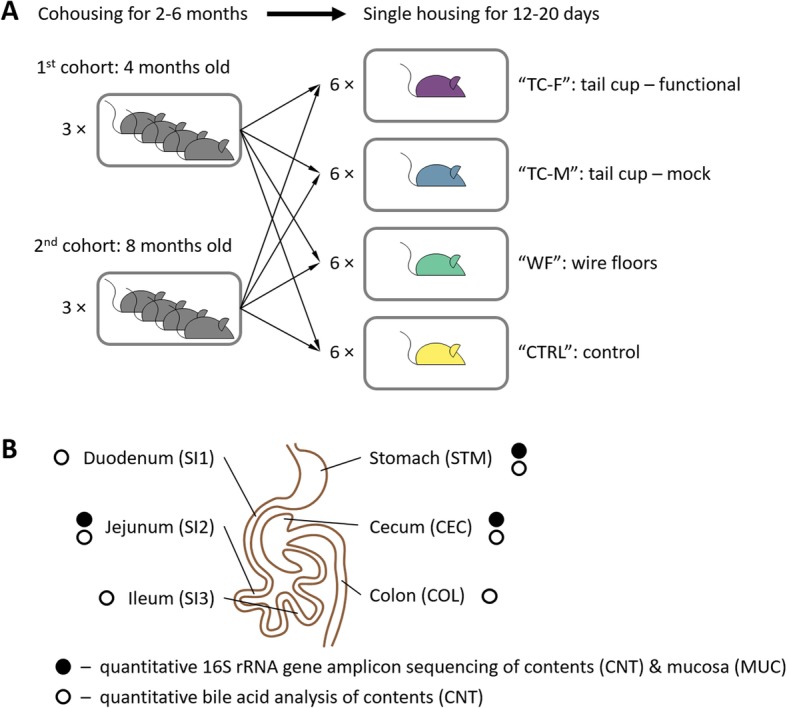
The upper gastrointestinal tract plays a prominent role in human physiology as the primary site for enzymatic digestion and nutrient absorption, immune sampling, and drug uptake. Alterations to the small intestine microbiome have been implicated in various human diseases, such as non-alcoholic steatohepatitis and inflammatory bowel conditions. Yet, the physiological and functional roles of the small intestine microbiota in humans remain poorly characterized because of the complexities associated with its sampling. Rodent models are used extensively in microbiome research and enable the spatial, temporal, compositional, and functional interrogation of the gastrointestinal microbiota and its effects on the host physiology and disease phenotype. Classical, culture-based studies have documented that fecal microbial self-reinoculation (via coprophagy) affects the composition and abundance of microbes in the murine proximal gastrointestinal tract. This pervasive self-reinoculation behavior could be a particularly relevant study factor when investigating small intestine microbiota. Modern microbiome studies either do not take self-reinoculation into account, or assume that approaches such as single housing mice or housing on wire mesh floors eliminate it. These assumptions have not been rigorously tested with modern tools. Here, we used quantitative 16S rRNA gene amplicon sequencing, quantitative microbial functional gene content inference, and metabolomic analyses of bile acids to evaluate the effects of self-reinoculation on microbial loads, composition, and function in the murine upper gastrointestinal tract.
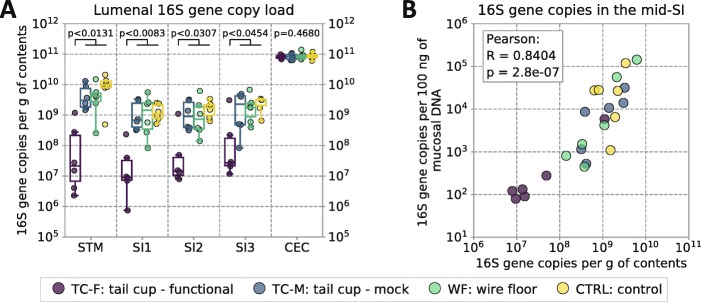
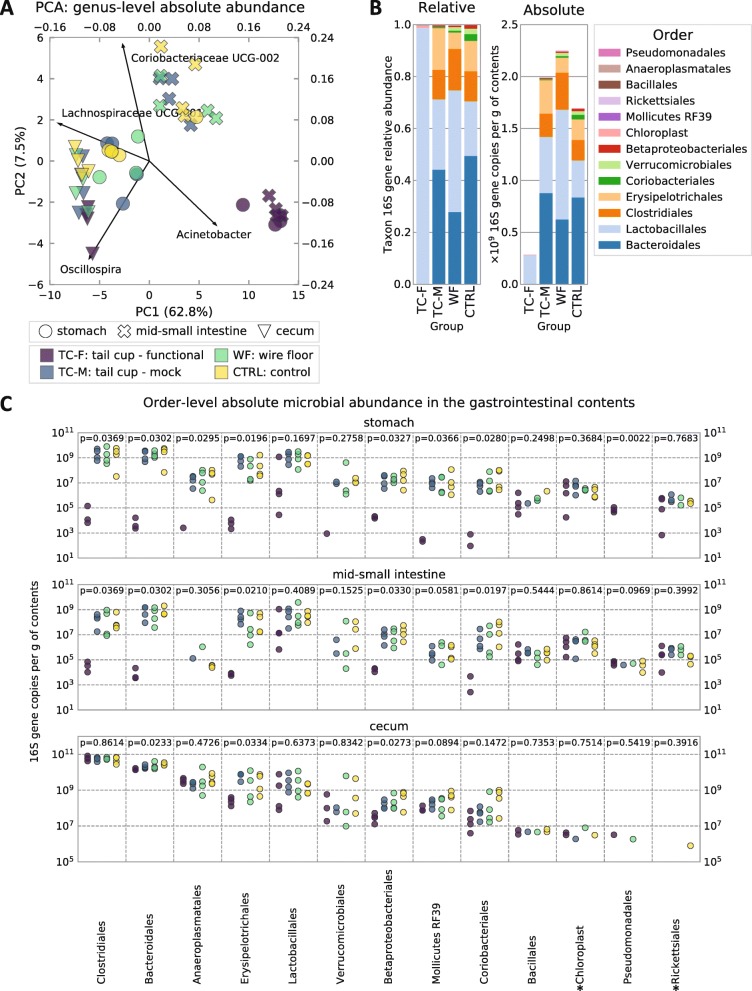
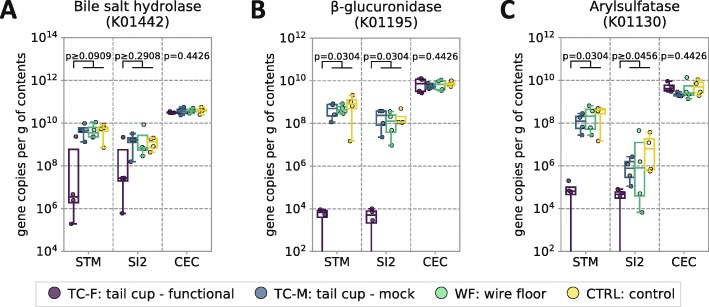
In coprophagic mice, continuous self-exposure to the fecal flora had substantial quantitative and qualitative effects on the upper gastrointestinal microbiome. These differences in microbial abundance and community composition were associated with an altered profile of the small intestine bile acid pool, and, importantly, could not be inferred from analyzing large intestine or stool samples. Overall, the patterns observed in the small intestine of non-coprophagic mice (reduced total microbial load, low abundance of anaerobic microbiota, and bile acids predominantly in the conjugated form) resemble those typically seen in the human small intestine.
Future studies need to take self-reinoculation into account when using mouse models to evaluate gastrointestinal microbial colonization and function in relation to xenobiotic transformation and pharmacokinetics or in the context of physiological states and diseases linked to small intestine microbiome and to small intestine dysbiosis. Video abstract.
上消化道在人类生理学中起着重要作用,是酶消化和营养吸收、免疫取样和药物摄取的主要场所。小肠微生物组的改变与各种人类疾病有关,如非酒精性脂肪性肝炎和炎症性肠病。然而,由于其采样的复杂性,小肠微生物组在人类中的生理和功能作用仍未得到很好的描述。啮齿动物模型在微生物组研究中被广泛应用,能够对胃肠道微生物组及其对宿主生理和疾病表型的影响进行空间、时间、组成和功能的研究。经典的、基于培养的研究已经记录了粪便微生物的自我再接种(通过食粪)影响了鼠类近端胃肠道中微生物的组成和丰度。这种普遍的自我再接种行为在研究小肠微生物组时可能是一个特别相关的研究因素。现代微生物组研究要么没有考虑到自我再接种,要么假设例如单笼饲养小鼠或饲养在金属网地板上可以消除它。这些假设尚未用现代工具进行严格测试。在这里,我们使用定量 16S rRNA 基因扩增子测序、定量微生物功能基因含量推断和胆汁酸代谢组学分析来评估自我再接种对鼠类上消化道微生物负荷、组成和功能的影响。
在食粪的小鼠中,连续自我暴露于粪便菌群对上消化道微生物组有显著的定量和定性影响。这些微生物丰度和群落组成的差异与小肠胆汁酸池的改变有关,重要的是,不能从分析大肠或粪便样本中推断出来。总的来说,非食粪小鼠小肠中观察到的模式(总微生物负荷降低、厌氧微生物群落丰度低、胆汁酸主要以结合形式存在)类似于人类小肠中通常观察到的模式。
未来的研究需要在使用小鼠模型评估与异生物质转化和药代动力学相关的胃肠道微生物定植和功能,或在与小肠微生物组和小肠菌群失调相关的生理状态和疾病的背景下,考虑自我再接种的因素。