Xu Qianhui, Chen Shaohuai, Hu Yuanbo, Huang Wen
The Second Affiliated Hospital and Yuying Children's Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, 325000, China.
Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310009, China.
Cell Death Discov. 2021 Nov 3;7(1):331. doi: 10.1038/s41420-021-00663-1.
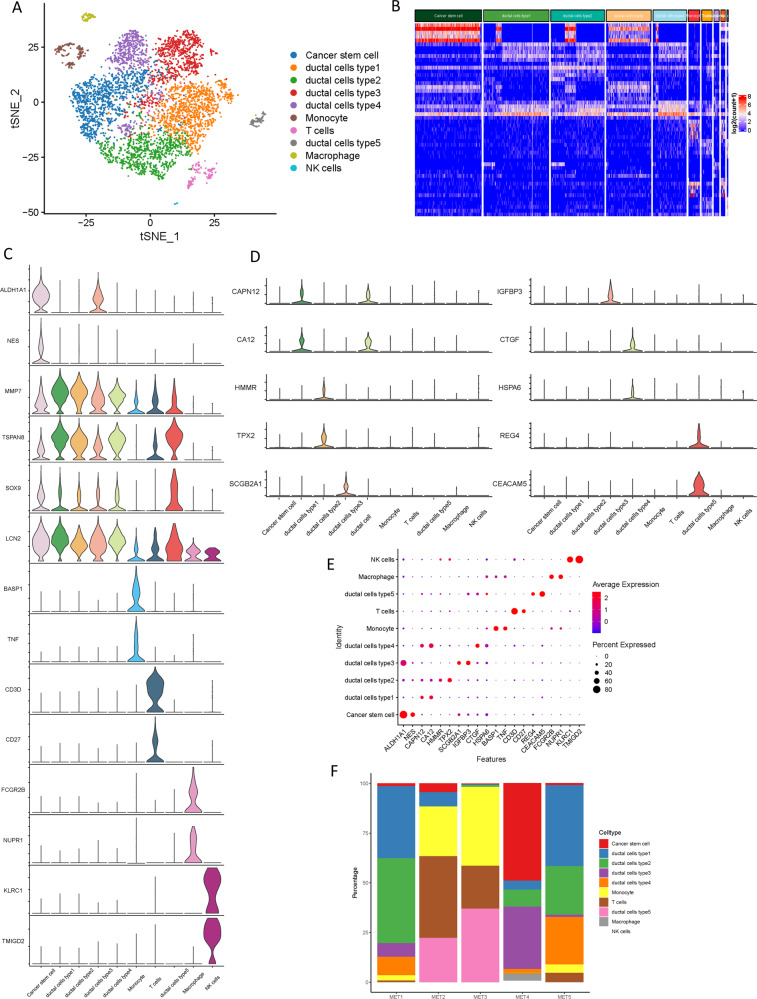
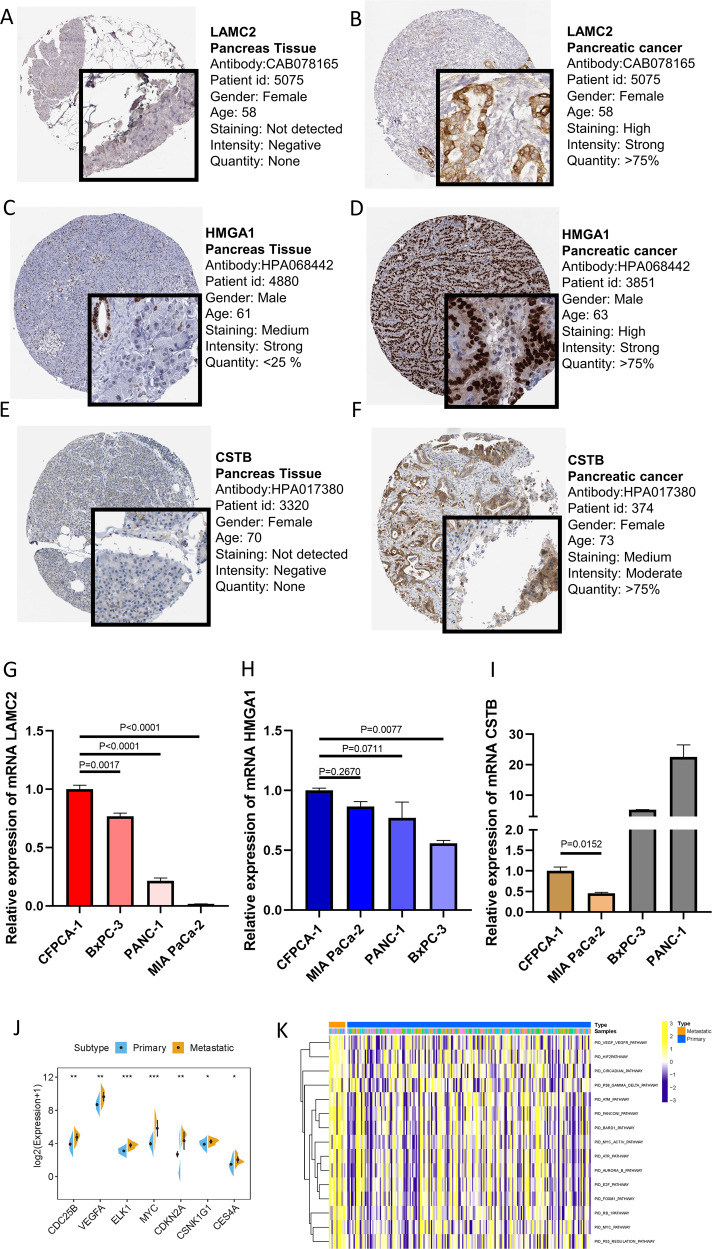
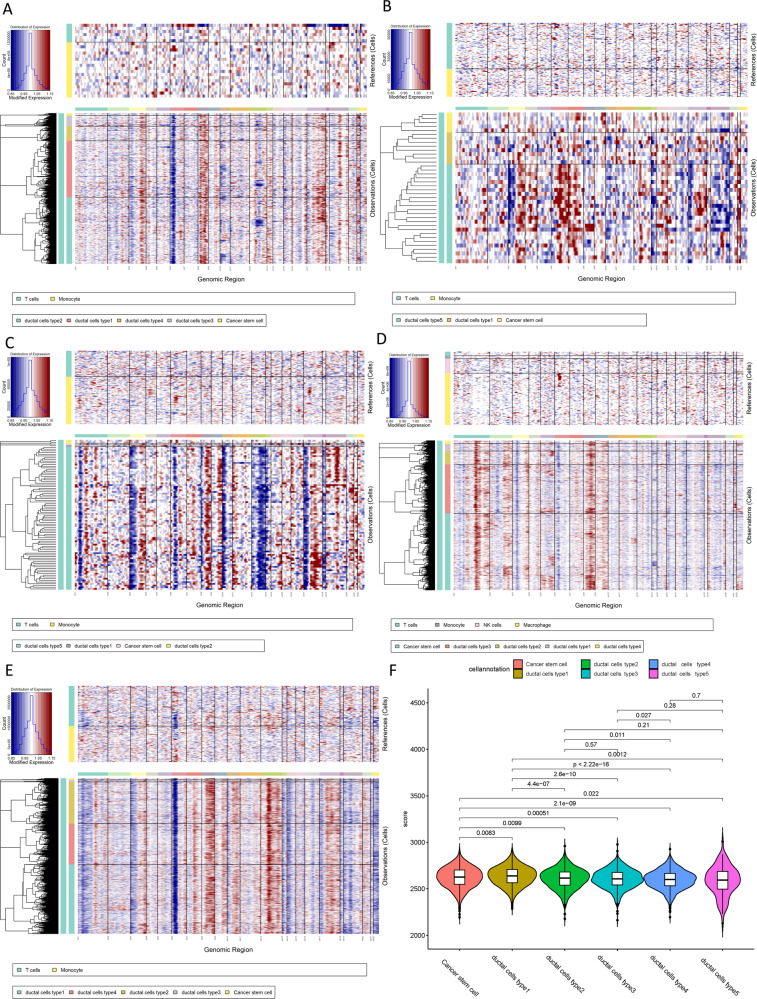
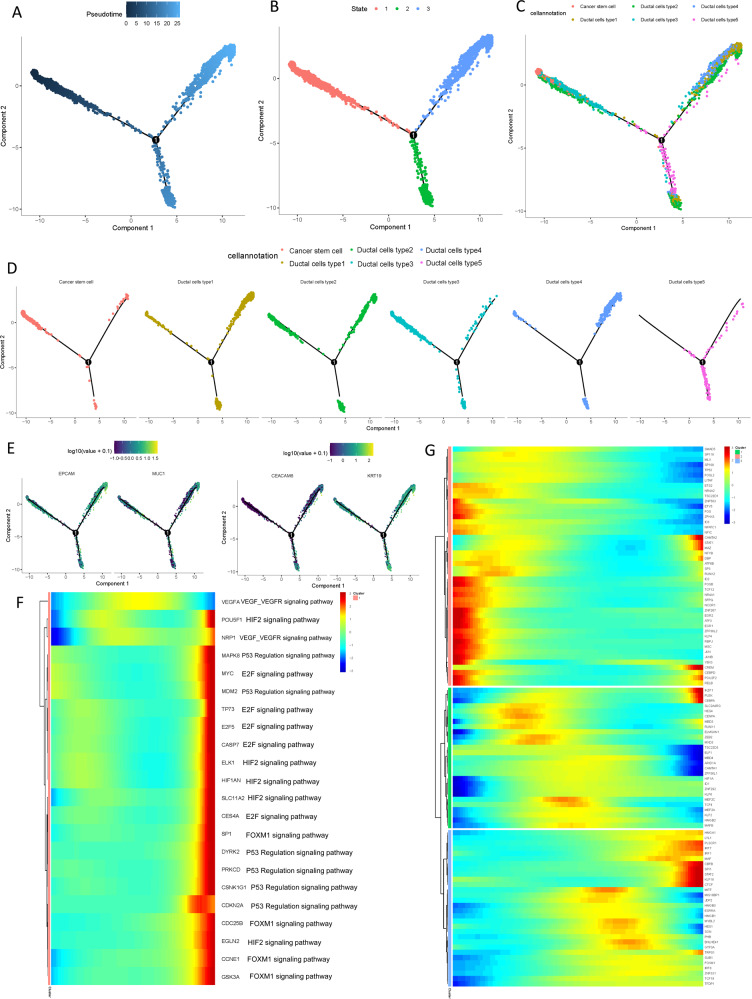
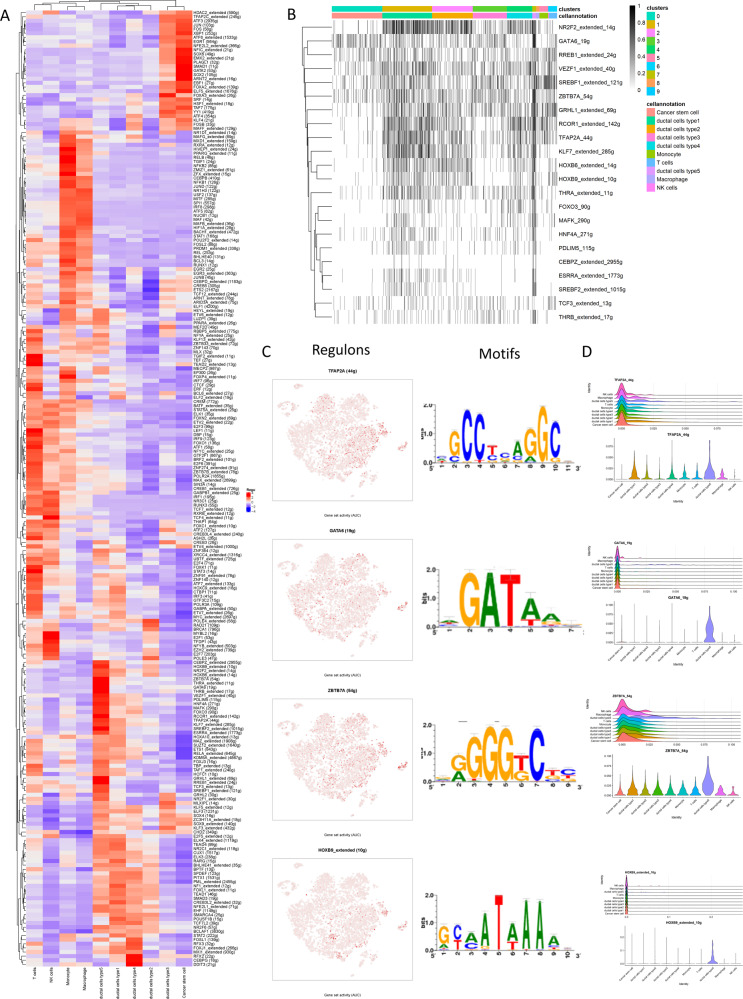
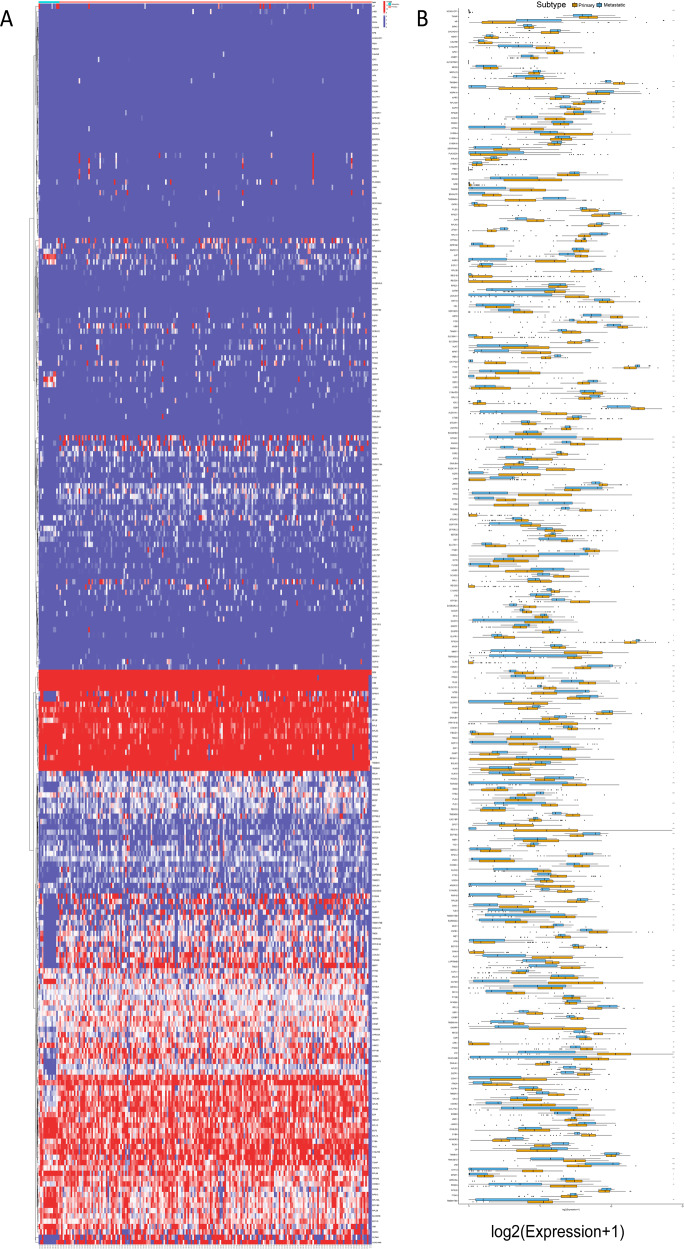
Pancreatic ductal adenocarcinoma (PDAC) is the most frequent and aggressive pancreatic tumor characterized by high metastatic risk and special tumor microenvironment. To comprehensively delineate the complex intra-tumoral heterogeneity and the underlying mechanism during metastatic lesions malignant progression, single-cell RNA sequencing (scRNA-seq) was employed. PCA and TSNE were used for dimensionality reduction analysis and cell clustering. Find All Markers function was used to calculate differential genes in each cluster, and Do Heatmap function was used to plot the distribution of differential genes in each cluster. GSVA was employed to assign pathway activity estimates to individual cells. Lineage trajectory progression was inferred by monocle. CNV status was inferred to compare the heterogeneity among patients and subtypes by infercnv. Ligand-receptor interactions were identified by CellPhoneDB, and regulons network of cells was analyzed by SCENIC. Through RNA-sequencing of 6236 individual cells from 5 liver metastatic PDAC lesions, 10 major cell clusters are identified by using unbiased clustering analysis of expression profiling and well-known cell markers. Cells with high CNV level were considered as malignant cells and pathway analyses were carried out to highlight intratumor heterogeneity in PDAC. Pseudotime trajectory analysis revealed that components of multiple tumor-related pathways and transcription factors (TFs) were differentially expressed along PDAC progression. The complex cellular communication suggested potential immunotherapeutic targets in PDAC. Regulon network identified multiple candidates for promising cell-specific transcriptional factors. Finally, metastatic-related genes expression levels and signaling pathways were validated in bulk RNA Sequencing data. This study contributed a comprehensive single-cell transcriptome atlas and contributed into novel insight of intratumor heterogeneity and molecular mechanism in metastatic PDAC.
胰腺导管腺癌(PDAC)是最常见且侵袭性最强的胰腺肿瘤,其特征为高转移风险和特殊的肿瘤微环境。为全面描绘转移性病变恶性进展过程中复杂的肿瘤内异质性及其潜在机制,采用了单细胞RNA测序(scRNA-seq)技术。主成分分析(PCA)和t分布随机邻域嵌入(TSNE)用于降维分析和细胞聚类。使用“查找所有标记”功能计算每个簇中的差异基因,并使用“绘制热图”功能绘制每个簇中差异基因的分布。基因集变异分析(GSVA)用于将通路活性估计值分配给单个细胞。通过单样本基因集轨迹推断(monocle)来推断谱系轨迹进展。通过infercnv推断拷贝数变异(CNV)状态,以比较患者和亚型之间的异质性。通过CellPhoneDB识别配体-受体相互作用,并通过单细胞调控网络推断与聚类(SCENIC)分析细胞的调控子网络。通过对来自5个肝转移性PDAC病变的6236个单个细胞进行RNA测序,利用表达谱的无偏聚类分析和知名细胞标记物鉴定出10个主要细胞簇。将具有高CNV水平的细胞视为恶性细胞,并进行通路分析以突出PDAC中的肿瘤内异质性。伪时间轨迹分析表明,多个肿瘤相关通路的成分和转录因子(TFs)在PDAC进展过程中差异表达。复杂的细胞通讯提示了PDAC中潜在的免疫治疗靶点。调控子网络鉴定出多个有前景的细胞特异性转录因子候选物。最后,在批量RNA测序数据中验证了转移相关基因的表达水平和信号通路。本研究贡献了一个全面的单细胞转录组图谱,并为转移性PDAC中的肿瘤内异质性和分子机制提供了新的见解。