Research Group in Computational Biology and Microbial Ecology, Department of Biological Sciences, Universidad de los Andes, Bogotá, Colombia.
Max Planck Tandem Group in Computational Biology, Universidad de los Andes, Bogotá, Colombia.
BMC Genomics. 2021 Nov 5;22(1):795. doi: 10.1186/s12864-021-08079-y.
Pathogens of the genus Phytophthora are the etiological agents of many devastating diseases in several high-value crops and forestry species such as potato, tomato, cocoa, and oak, among many others. Phytophthora betacei is a recently described species that causes late blight almost exclusively in tree tomatoes, and it is closely related to Phytophthora infestans that causes the disease in potato crops and other Solanaceae. This study reports the assembly and annotation of the genomes of P. betacei P8084, the first of its species, and P. infestans RC1-10, a Colombian strain from the EC-1 lineage, using long-read SMRT sequencing technology.
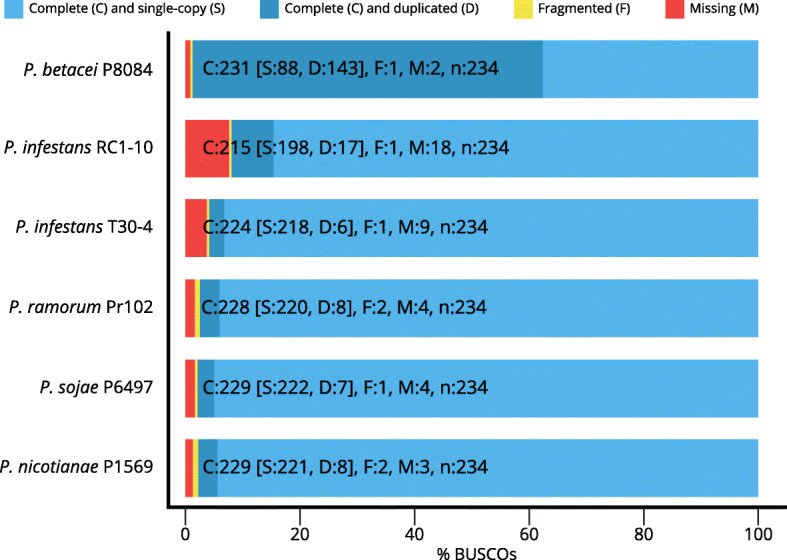
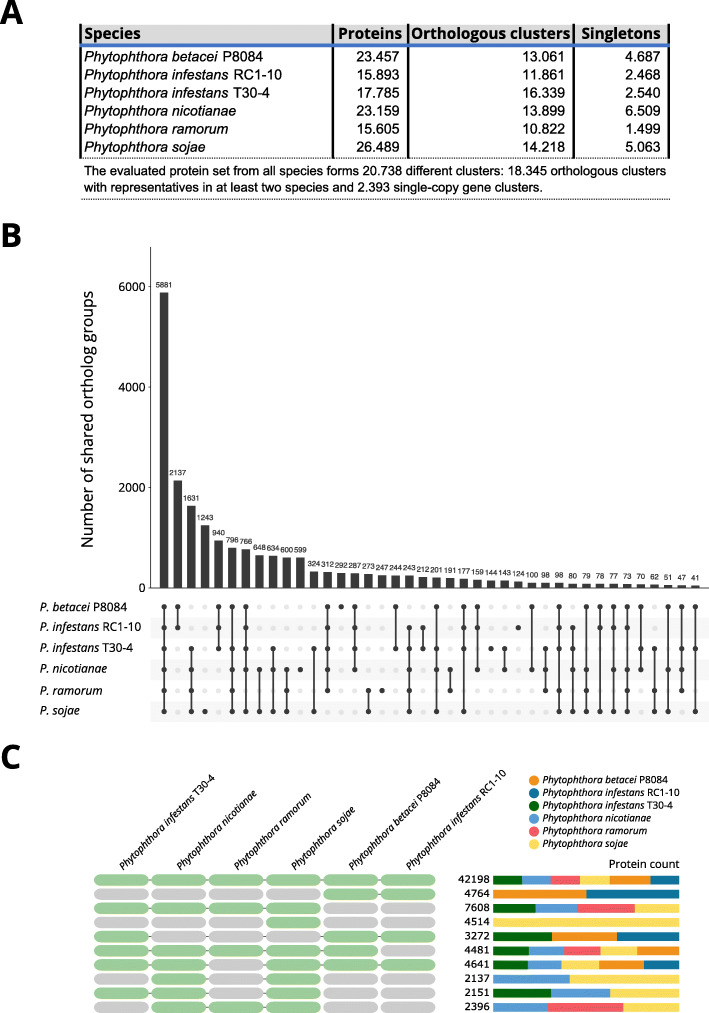
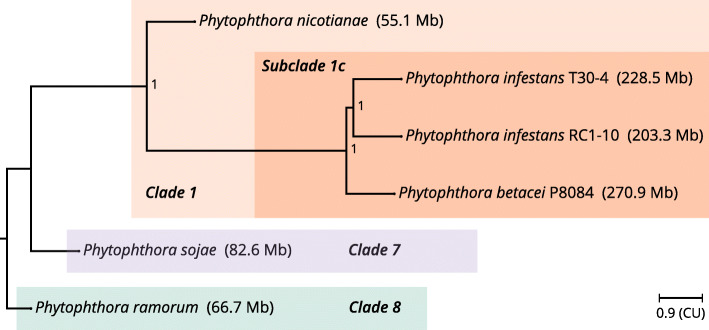
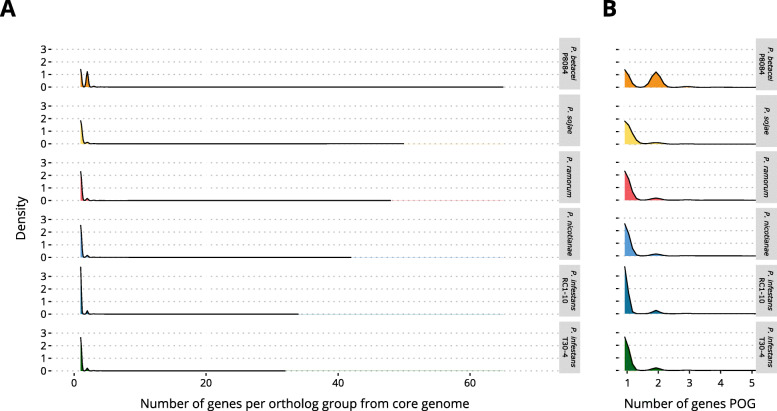
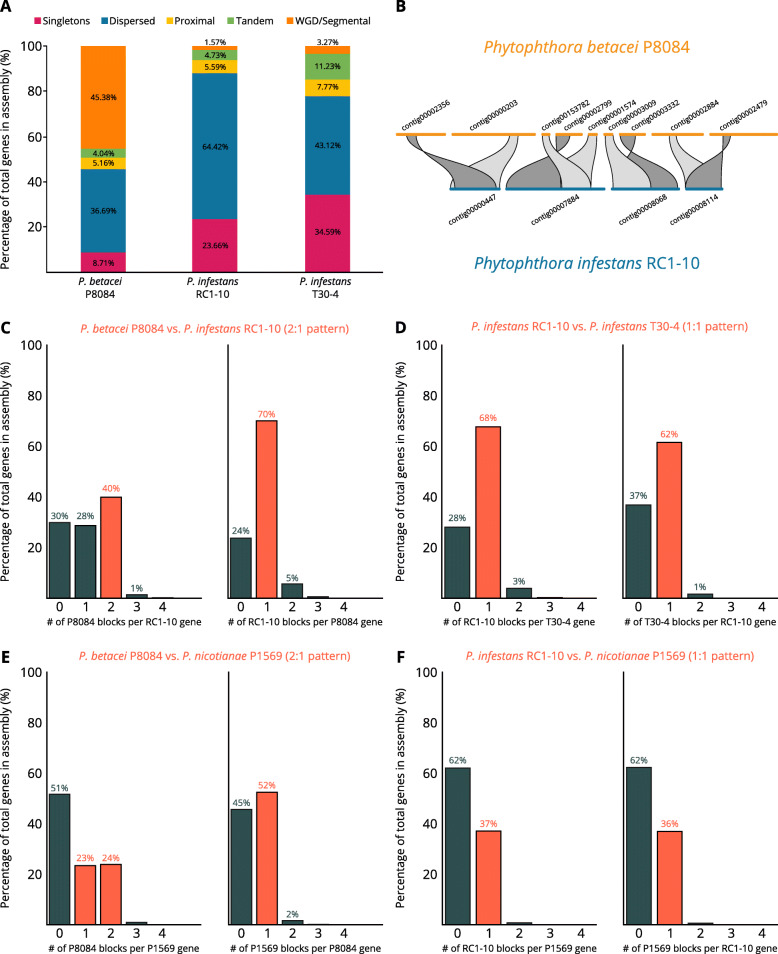
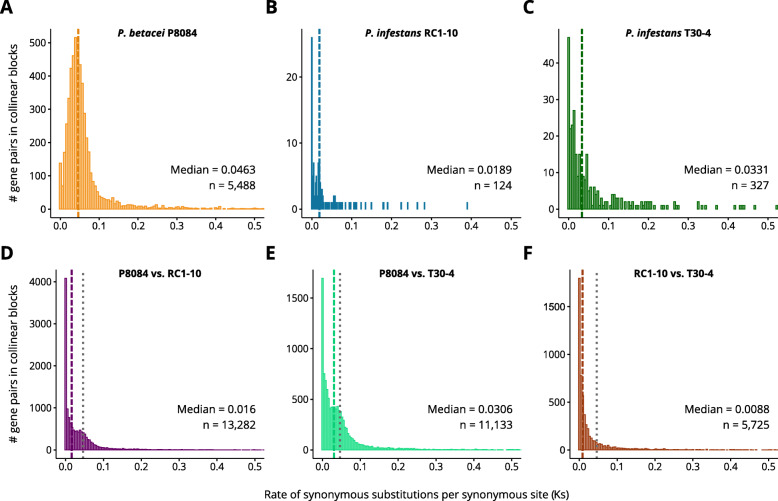
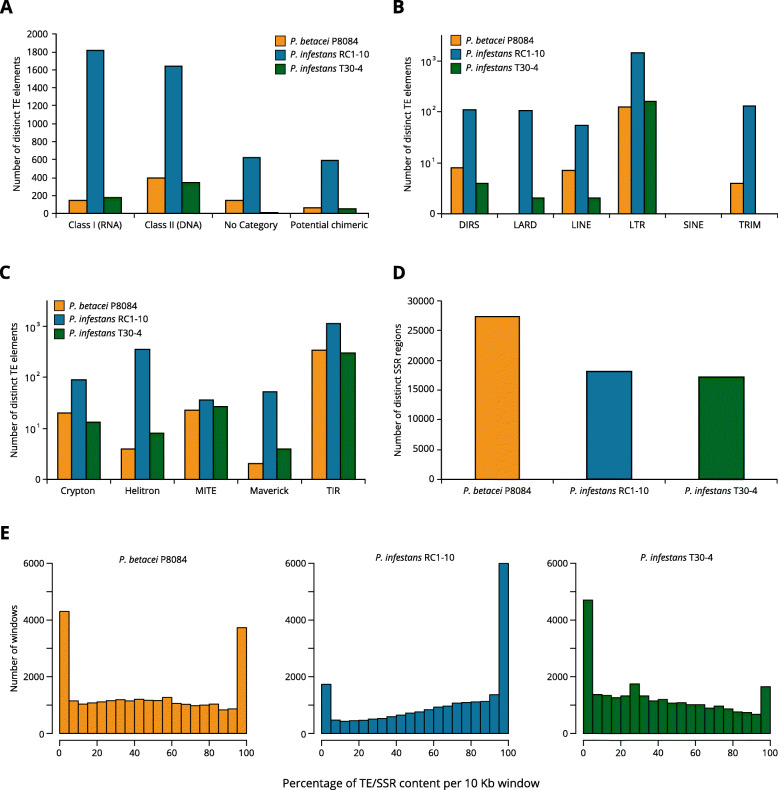
Our results show that P. betacei has the largest sequenced genome size of the Phytophthora genus so far with 270 Mb. A moderate transposable element invasion and a whole genome duplication likely explain its genome size expansion when compared to P. infestans, whereas P. infestans RC1-10 has expanded its genome under the activity of transposable elements. The high diversity and abundance (in terms of copy number) of classified and unclassified transposable elements in P. infestans RC1-10 relative to P. betacei bears testimony of the power of long-read technologies to discover novel repetitive elements in the genomes of organisms. Our data also provides support for the phylogenetic placement of P. betacei as a standalone species and as a sister group of P. infestans. Finally, we found no evidence to support the idea that the genome of P. betacei P8084 follows the same gene-dense/gense-sparse architecture proposed for P. infestans and other filamentous plant pathogens.
This study provides the first genome-wide picture of P. betacei and expands the genomic resources available for P. infestans. This is a contribution towards the understanding of the genome biology and evolutionary history of Phytophthora species belonging to the subclade 1c.
腐霉属的病原体是许多高价值作物和林业物种(如马铃薯、番茄、可可和橡树等)多种毁灭性疾病的病原体。Phytophthora betacei 是最近描述的一种物种,它几乎只在树番茄中引起晚疫病,与引起马铃薯作物和其他茄科植物病害的 Phytophthora infestans 密切相关。本研究报告了第一个 P. betacei P8084 及其种的基因组组装和注释,以及 P. infestans RC1-10,这是一个来自 EC-1 谱系的哥伦比亚菌株,使用长读 SMRT 测序技术。
我们的结果表明,与 P. infestans 相比,P. betacei 具有迄今为止腐霉属最大的测序基因组大小,为 270 Mb。中等程度的转座子入侵和全基因组复制可能解释了其基因组大小的扩张,而 P. infestans RC1-10 则在转座子的作用下扩展了其基因组。与 P. betacei 相比,P. infestans RC1-10 中分类和未分类转座子的高多样性和丰富度(以拷贝数计)证明了长读技术在发现生物体基因组中新型重复元件方面的强大功能。我们的数据还支持 P. betacei 作为一个独立物种和 P. infestans 的姐妹群的系统发育位置。最后,我们没有发现任何证据支持 P. betacei P8084 的基因组遵循与 P. infestans 和其他丝状植物病原体提出的相同基因密集/稀疏结构的想法。
本研究提供了 P. betacei 的全基因组图谱,并扩展了 P. infestans 可用的基因组资源。这是对属于亚群 1c 的腐霉属物种的基因组生物学和进化历史的理解的贡献。