Systems and Computing Engineering Department, Universidad de los Andes, Bogotá 111711, Colombia.
Biotechnology lab, Centro de Investigación de la caña de azúcar de Colombia, CENICAÑA, Cali 760046, Colombia.
Bioinformatics. 2019 Nov 1;35(22):4716-4723. doi: 10.1093/bioinformatics/btz275.
Accurate detection, genotyping and downstream analysis of genomic variants from high-throughput sequencing data are fundamental features in modern production pipelines for genetic-based diagnosis in medicine or genomic selection in plant and animal breeding. Our research group maintains the Next-Generation Sequencing Experience Platform (NGSEP) as a precise, efficient and easy-to-use software solution for these features.
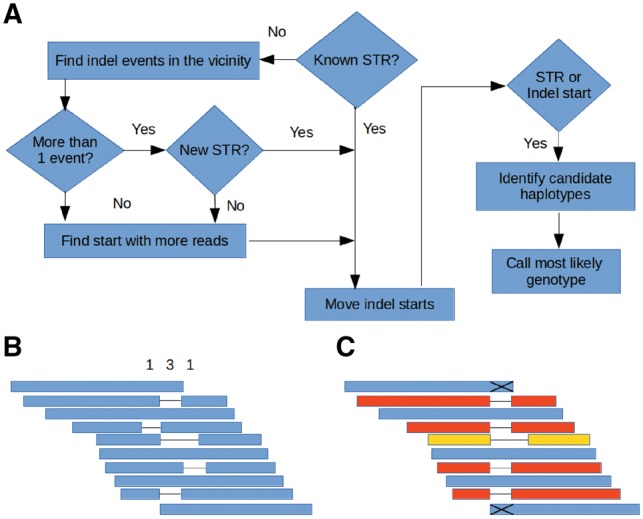
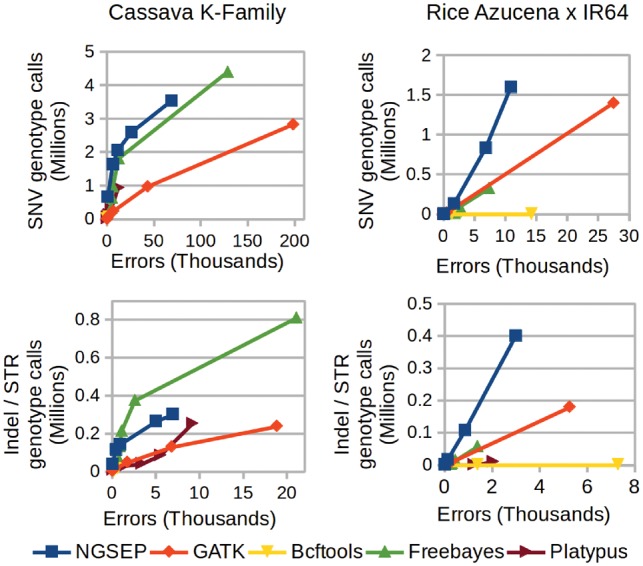
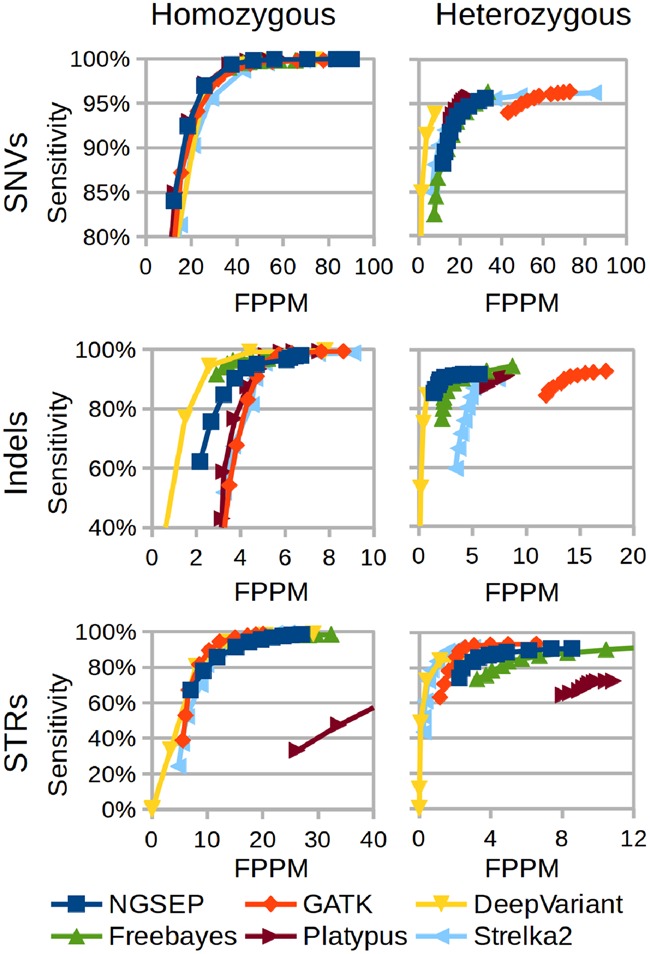
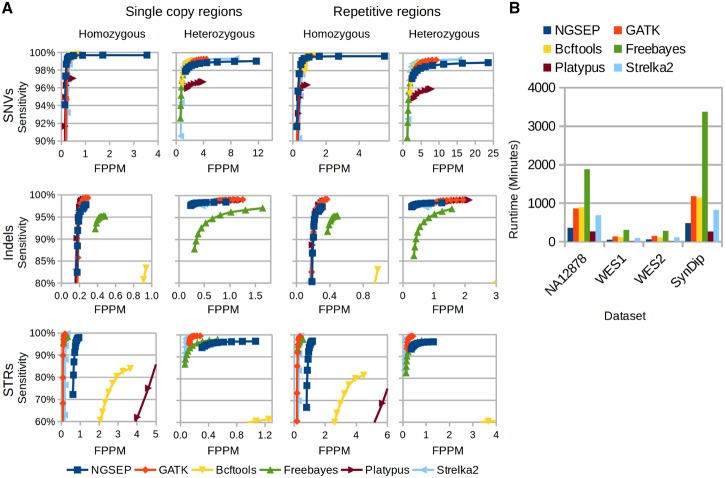
Understanding that incorrect alignments around short tandem repeats are an important source of genotyping errors, we implemented in NGSEP new algorithms for realignment and haplotype clustering of reads spanning indels and short tandem repeats. We performed extensive benchmark experiments comparing NGSEP to state-of-the-art software using real data from three sequencing protocols and four species with different distributions of repetitive elements. NGSEP consistently shows comparative accuracy and better efficiency compared to the existing solutions. We expect that this work will contribute to the continuous improvement of quality in variant calling needed for modern applications in medicine and agriculture.
NGSEP is available as open source software at http://ngsep.sf.net.
Supplementary data are available at Bioinformatics online.
从高通量测序数据中准确检测、基因分型和下游分析基因组变异是医学中基于遗传的诊断或植物和动物育种中基于基因组选择的现代生产管道中的基本特征。我们的研究小组维护着下一代测序体验平台(NGSEP),作为一种精确、高效和易于使用的软件解决方案,具有这些功能。
我们认识到短串联重复周围不正确的比对是基因分型错误的一个重要来源,因此在 NGSEP 中实现了新的算法,用于重新比对和单倍型聚类跨越插入缺失和短串联重复的读取。我们使用来自三种测序方案和四种具有不同重复元件分布的物种的真实数据,对 NGSEP 与最先进的软件进行了广泛的基准测试实验。与现有解决方案相比,NGSEP 始终表现出相当的准确性和更好的效率。我们期望这项工作将有助于不断提高医学和农业现代应用中所需的变异调用质量。
NGSEP 可在 http://ngsep.sf.net 上作为开源软件获得。
补充数据可在生物信息学在线获得。