Institute of Medical and Human Genetics, Charité Universitätsmedizin, Berlin, Germany.
Core Unit Bioinformatics, Berlin Institute of Health, Berlin, Germany.
Eur J Hum Genet. 2022 Feb;30(2):178-186. doi: 10.1038/s41431-021-00983-x. Epub 2021 Nov 8.
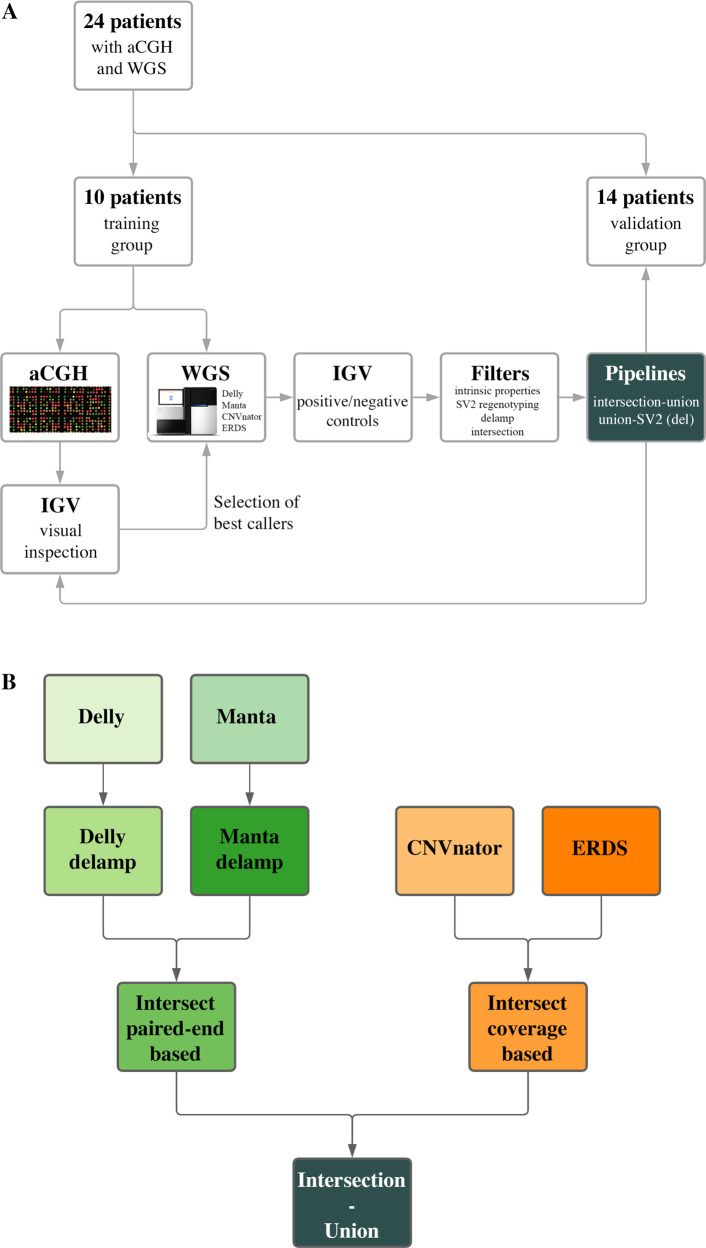
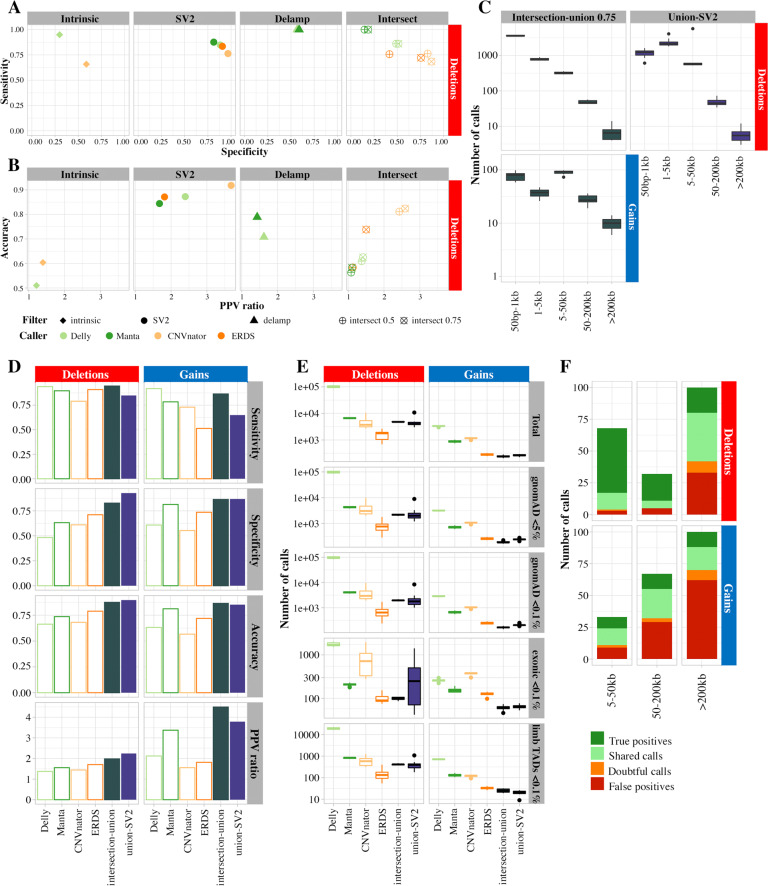
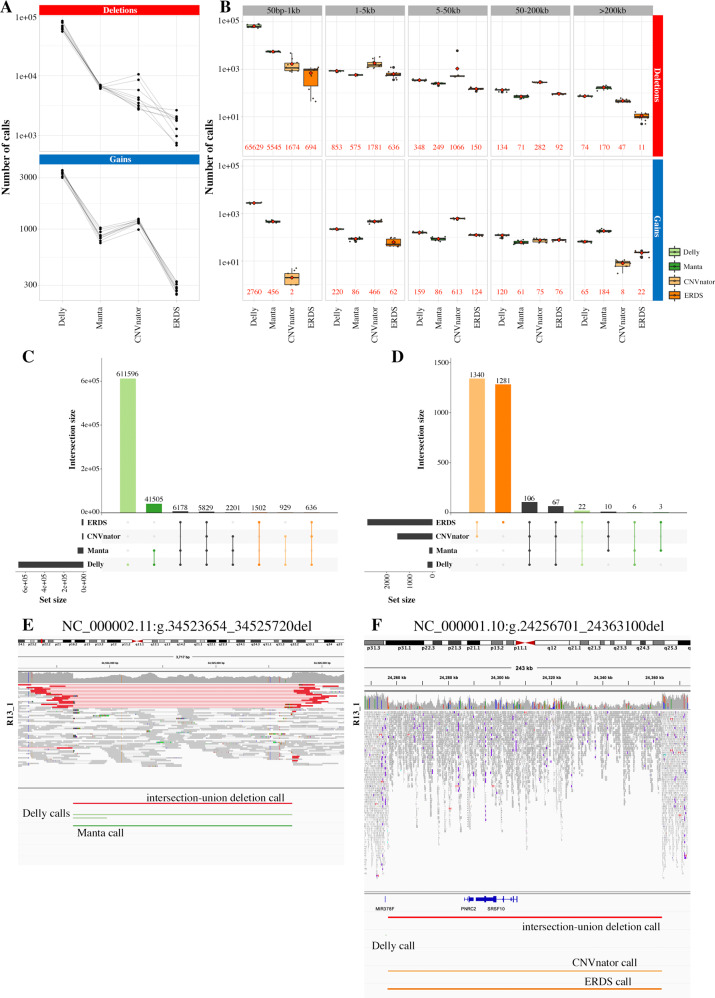
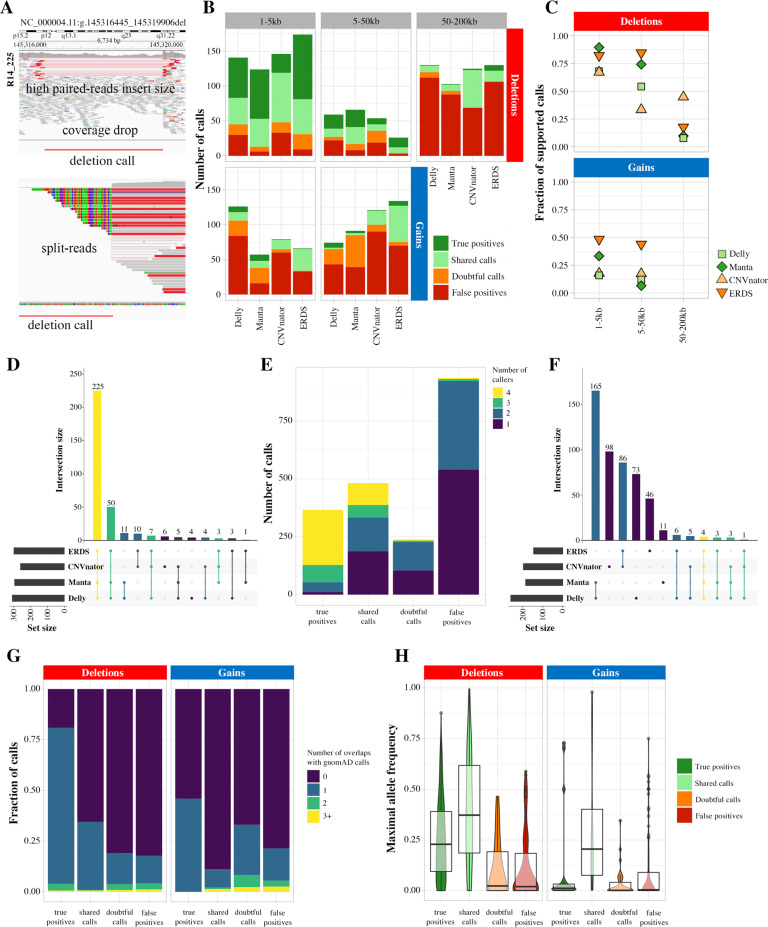
Copy Number Variants (CNVs) are deletions, duplications or insertions larger than 50 base pairs. They account for a large percentage of the normal genome variation and play major roles in human pathology. While array-based approaches have long been used to detect them in clinical practice, whole-genome sequencing (WGS) bears the promise to allow concomitant exploration of CNVs and smaller variants. However, accurately calling CNVs from WGS remains a difficult computational task, for which a consensus is still lacking. In this paper, we explore practical calling options to reach the best compromise between sensitivity and sensibility. We show that callers based on different signal (paired-end reads, split reads, coverage depth) yield complementary results. We suggest approaches combining four selected callers (Manta, Delly, ERDS, CNVnator) and a regenotyping tool (SV2), and show that this is applicable in everyday practice in terms of computation time and further interpretation. We demonstrate the superiority of these approaches over array-based Comparative Genomic Hybridization (aCGH), specifically regarding the lack of resolution in breakpoint definition and the detection of potentially relevant CNVs. Finally, we confirm our results on the NA12878 benchmark genome, as well as one clinically validated sample. In conclusion, we suggest that WGS constitutes a timely and economically valid alternative to the combination of aCGH and whole-exome sequencing.
拷贝数变异(CNVs)是指 50 个碱基对以上的缺失、重复或插入。它们占正常基因组变异的很大比例,并在人类病理学中发挥重要作用。虽然基于阵列的方法长期以来一直用于临床实践中检测它们,但全基因组测序(WGS)有望同时探索 CNVs 和较小的变体。然而,从 WGS 中准确地调用 CNVs 仍然是一项具有挑战性的计算任务,目前仍缺乏共识。在本文中,我们探讨了实用的调用选项,以在灵敏度和特异性之间达到最佳折衷。我们表明,基于不同信号(成对读取、分裂读取、覆盖深度)的调用器产生互补的结果。我们提出了结合四个选定的调用器(Manta、Delly、ERDS、CNVnator)和一个重分型工具(SV2)的方法,并表明在计算时间和进一步解释方面,这在日常实践中是可行的。我们证明了这些方法优于基于阵列的比较基因组杂交(aCGH),特别是在定义断点的分辨率和检测潜在相关 CNVs 方面。最后,我们在 NA12878 基准基因组和一个临床验证样本上验证了我们的结果。总之,我们认为 WGS 是 aCGH 和全外显子组测序组合的及时且经济有效的替代方案。