MGI, BGI-Shenzhen, Shenzhen, 518083, China.
BGI-Shenzhen, Shenzhen, 518083, China.
BMC Bioinformatics. 2020 Nov 11;21(1):518. doi: 10.1186/s12859-020-03859-x.
DNBSEQ™ platforms are new massively parallel sequencing (MPS) platforms that use DNA nanoball technology. Use of data generated from DNBSEQ™ platforms to detect single nucleotide variants (SNVs) and small insertions and deletions (indels) has proven to be quite effective, while the feasibility of copy number variants (CNVs) detection is unclear.
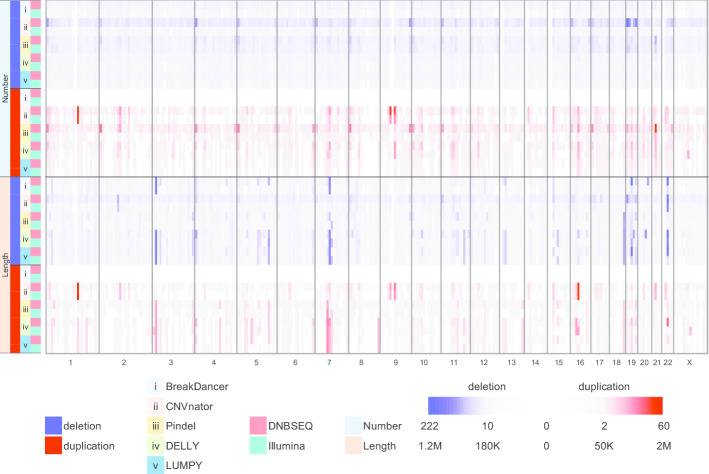
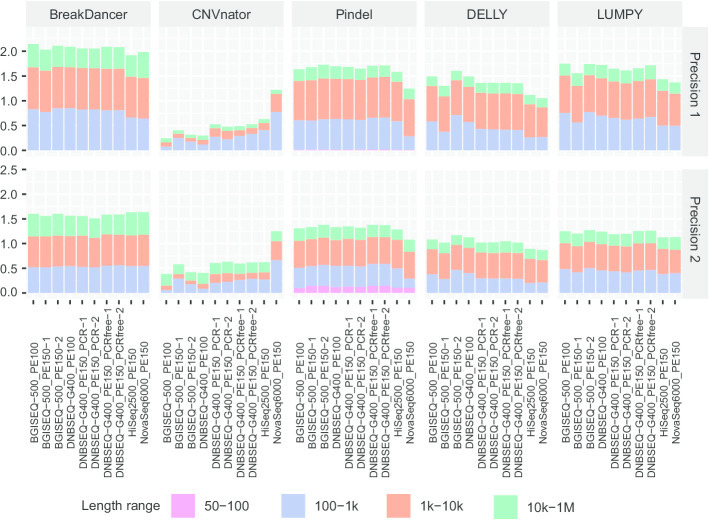
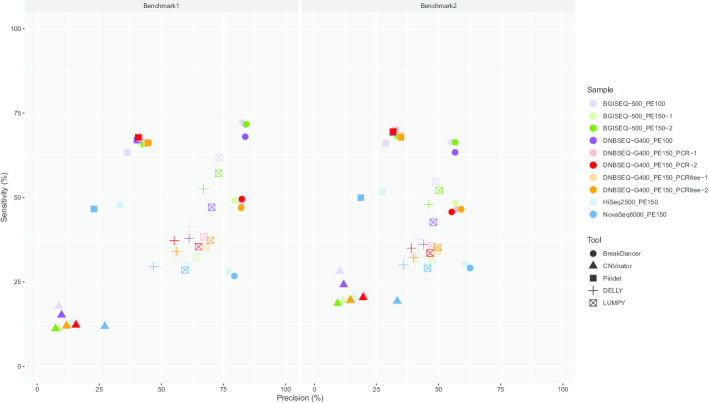
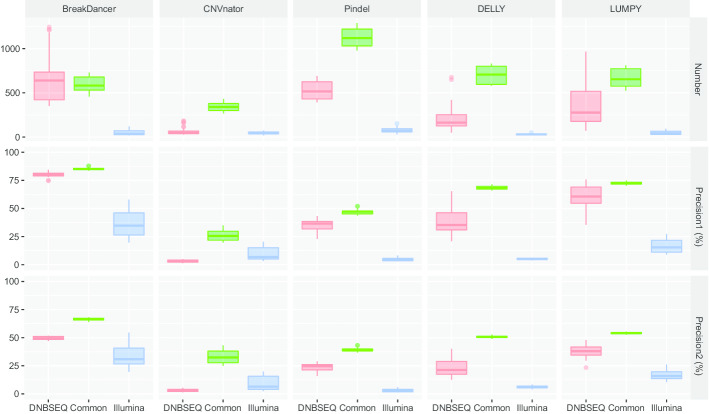
Here, we first benchmarked different CNV detection tools based on Illumina whole-genome sequencing (WGS) data of NA12878 and then assessed these tools in CNV detection based on DNBSEQ™ sequencing data from the same sample. When the same tool was used, the CNVs detected based on DNBSEQ™ and Illumina data were similar in quantity, length and distribution, while great differences existed within results from different tools and even based on data from a single platform. We further estimated the CNV detection power based on available CNV benchmarks of NA12878 and found similar precision and sensitivity between the DNBSEQ™ and Illumina platforms. We also found higher precision of CNVs shorter than 1 kbp based on DNBSEQ™ platforms than those based on Illumina platforms by using Pindel, DELLY and LUMPY. We carefully compared these two available benchmarks and found a large proportion of specific CNVs between them. Thus, we constructed a more complete CNV benchmark of NA12878 containing 3512 CNV regions.
We assessed and benchmarked CNV detections based on WGS with DNBSEQ™ platforms and provide guidelines for future studies.
DNBSEQ™ 平台是使用 DNA 纳米球技术的新型大规模平行测序(MPS)平台。使用 DNBSEQ™ 平台生成的数据来检测单核苷酸变异(SNVs)和小插入和缺失(indels)已被证明非常有效,而检测拷贝数变异(CNVs)的可行性尚不清楚。
在这里,我们首先基于 Illumina 全基因组测序(WGS)数据对不同的 CNV 检测工具进行了基准测试,然后评估了这些工具在基于相同样本的 DNBSEQ™ 测序数据的 CNV 检测中的性能。当使用相同的工具时,基于 DNBSEQ™ 和 Illumina 数据检测到的 CNVs 在数量、长度和分布上相似,而不同工具的结果之间以及基于单个平台的数据之间存在很大差异。我们进一步根据 NA12878 的可用 CNV 基准评估了 CNV 检测能力,发现 DNBSEQ™ 和 Illumina 平台之间的精度和灵敏度相似。我们还发现,基于 DNBSEQ™ 平台的短于 1 kbp 的 CNVs 的精度高于基于 Illumina 平台的精度,这是通过 Pindel、DELLY 和 LUMPY 实现的。我们仔细比较了这两个可用的基准,并发现它们之间存在很大比例的特定 CNVs。因此,我们构建了一个包含 3512 个 CNV 区域的更完整的 NA12878 的 CNV 基准。
我们评估和基准测试了基于 DNBSEQ™ 平台的 WGS 中的 CNV 检测,并为未来的研究提供了指导。