Kovacs Gergo, Reimer Lasse, Jensen Poul Henning
Danish Research Institute of Translational Neuroscience - DANDRITE, Aarhus University, Aarhus, Denmark.
Department of Biomedicine, Aarhus University, Aarhus, Denmark.
Front Neurol. 2021 Oct 20;12:742625. doi: 10.3389/fneur.2021.742625. eCollection 2021.
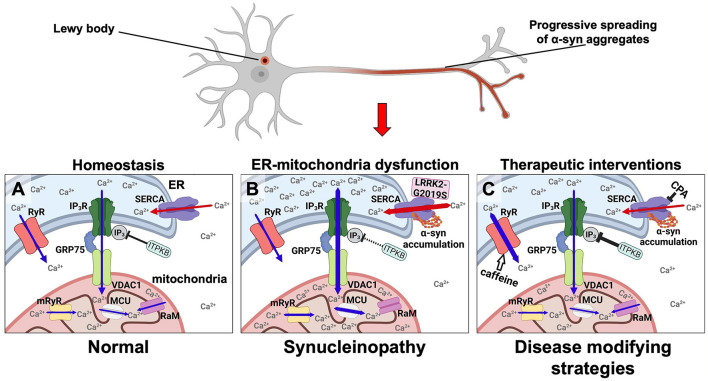
Neuronal calcium dyshomeostasis has been associated to Parkinson's disease (PD) development based on epidemiological studies on users of calcium channel antagonists and clinical trials are currently conducted exploring the hypothesis of increased calcium influx into neuronal cytosol as basic premise. We reported in 2018 an opposite hypothesis based on the demonstration that α-synuclein aggregates stimulate the endoplasmic reticulum (ER) calcium pump SERCA and demonstrated in cell models the existence of an α-synuclein-aggregate dependent neuronal state wherein cytosolic calcium is decreased due to an increased pumping of calcium into the ER. Inhibiting the SERCA pump protected both neurons and an α-synuclein transgenic model. This models two cellular states that could contribute to development of PD. First the prolonged state with reduced cytosolic calcium that could deregulate multiple signaling pathways. Second the disease ER state with increased calcium concentration. We will discuss our hypothesis in the light of recent papers. First, a mechanistic study describing how variation in the Inositol-1,4,5-triphosphate (IP3) kinase B (ITPKB) may explain GWAS studies identifying the ITPKB gene as a protective factor toward PD. Here it was demonstrated that how increased ITPKB activity reduces influx of ER calcium to mitochondria via contact between IP-receptors and the mitochondrial calcium uniporter complex in ER-mitochondria contact, known as mitochondria-associated membranes (MAMs). Secondly, it was demonstrated that astrocytes derived from PD patients contain α-synuclein accumulations. A recent study has demonstrated how human astrocytes derived from a few PD patients carrying the LRRK2-2019S mutation express more α-synuclein than control astrocytes, release more calcium from ER upon ryanodine receptor (RyR) stimulation, show changes in ER calcium channels and exhibit a decreased maximal and spare respiration indicating altered mitochondrial function in PD astrocytes. Here, we summarize the previous findings focusing the effect of α-synuclein to SERCA, RyR, IPR, MCU subunits and other MAM-related channels. We also consider how the SOCE-related events could contribute to the development of PD.
基于对钙通道拮抗剂使用者的流行病学研究,神经元钙稳态失衡与帕金森病(PD)的发展有关,目前正在进行临床试验,以探索钙流入神经元细胞质增加这一假说作为基本前提。我们在2018年报道了一个相反的假说,该假说基于α-突触核蛋白聚集体刺激内质网(ER)钙泵SERCA的证明,并在细胞模型中证明了存在一种α-突触核蛋白聚集体依赖性神经元状态,其中由于钙向内质网的泵送增加,细胞质钙减少。抑制SERCA泵对神经元和α-突触核蛋白转基因模型均有保护作用。这模拟了两种可能导致PD发展的细胞状态。第一种是细胞质钙减少的延长状态,这可能会使多种信号通路失调。第二种是钙浓度增加的疾病内质网状态。我们将根据最近的论文讨论我们的假说。首先,一项机制研究描述了肌醇-1,4,5-三磷酸(IP3)激酶B(ITPKB)的变异如何解释全基因组关联研究(GWAS)将ITPKB基因鉴定为PD的保护因子。该研究表明,ITPKB活性增加如何通过内质网-线粒体接触(称为线粒体相关膜,MAMs)中IP受体与线粒体钙单向转运体复合物之间的接触减少内质网钙向线粒体的流入。其次,已证明源自PD患者的星形胶质细胞含有α-突触核蛋白聚集物。最近的一项研究表明,来自少数携带LRRK2-2019S突变的PD患者的人星形胶质细胞比对照星形胶质细胞表达更多的α-突触核蛋白,在ryanodine受体(RyR)刺激后从内质网释放更多的钙,内质网钙通道发生变化,并表现出最大呼吸和备用呼吸降低,表明PD星形胶质细胞中的线粒体功能改变。在此,我们总结先前的发现,重点关注α-突触核蛋白对SERCA、RyR、IPR、MCU亚基和其他与MAM相关通道的影响。我们还考虑了与钙库操纵性钙内流(SOCE)相关的事件如何可能导致PD的发展。