Sheik Amamuddy Olivier, Afriyie Boateng Rita, Barozi Victor, Wavinya Nyamai Dorothy, Tastan Bishop Özlem
Research Unit in Bioinformatics (RUBi), Department of Biochemistry and Microbiology, Rhodes University, Makhanda, South Africa.
Comput Struct Biotechnol J. 2021;19:6431-6455. doi: 10.1016/j.csbj.2021.11.016. Epub 2021 Nov 25.
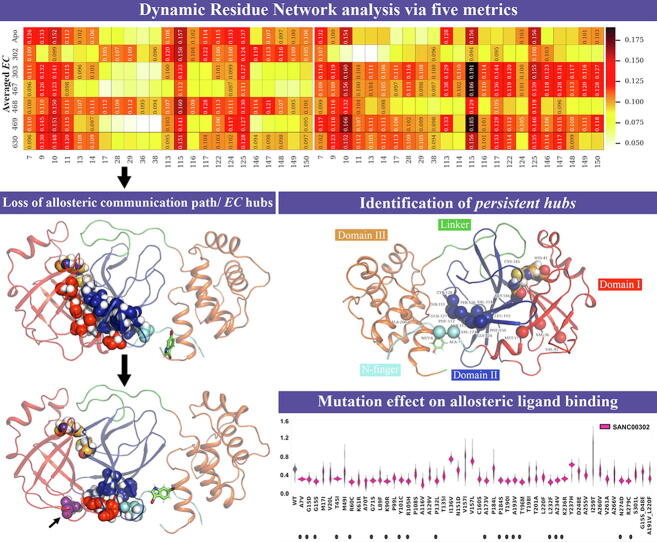
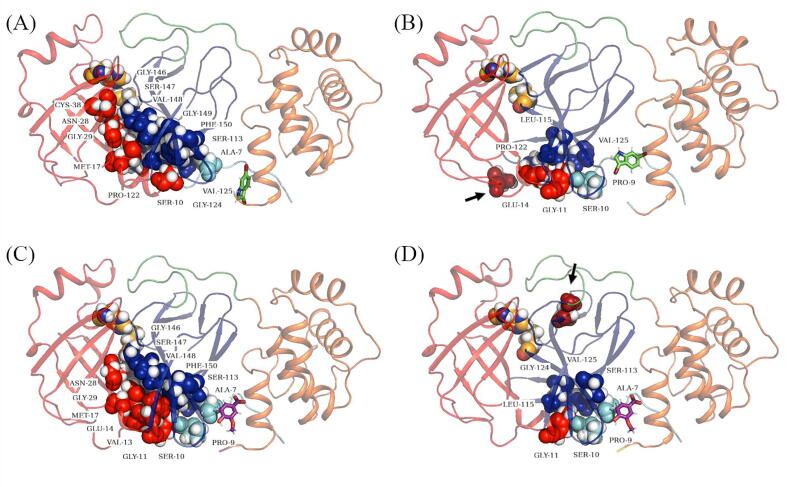
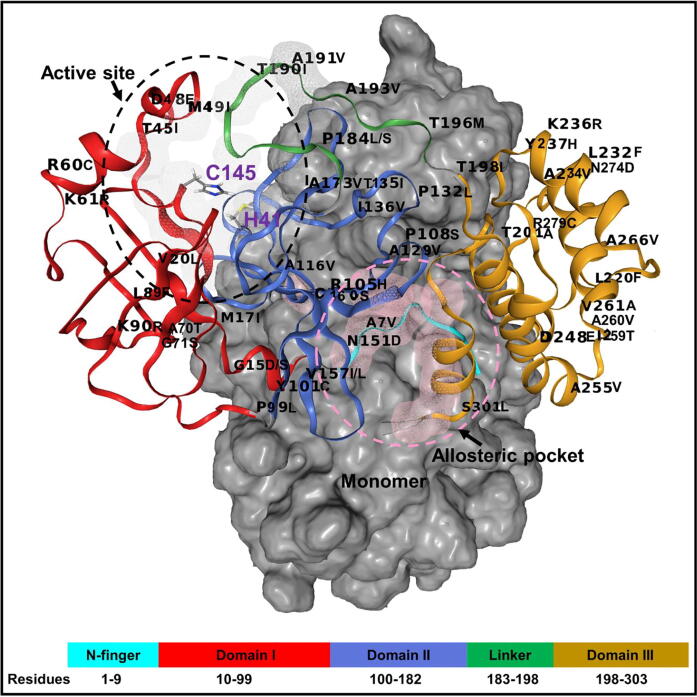
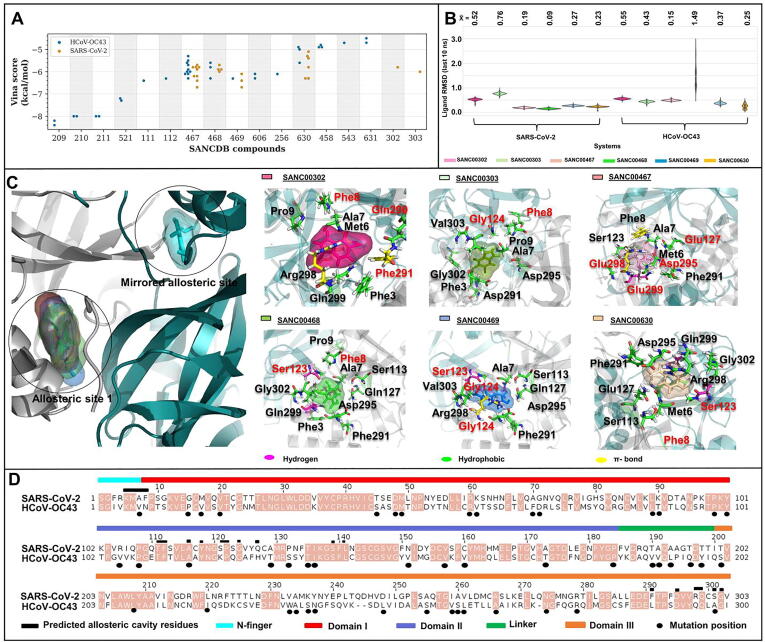
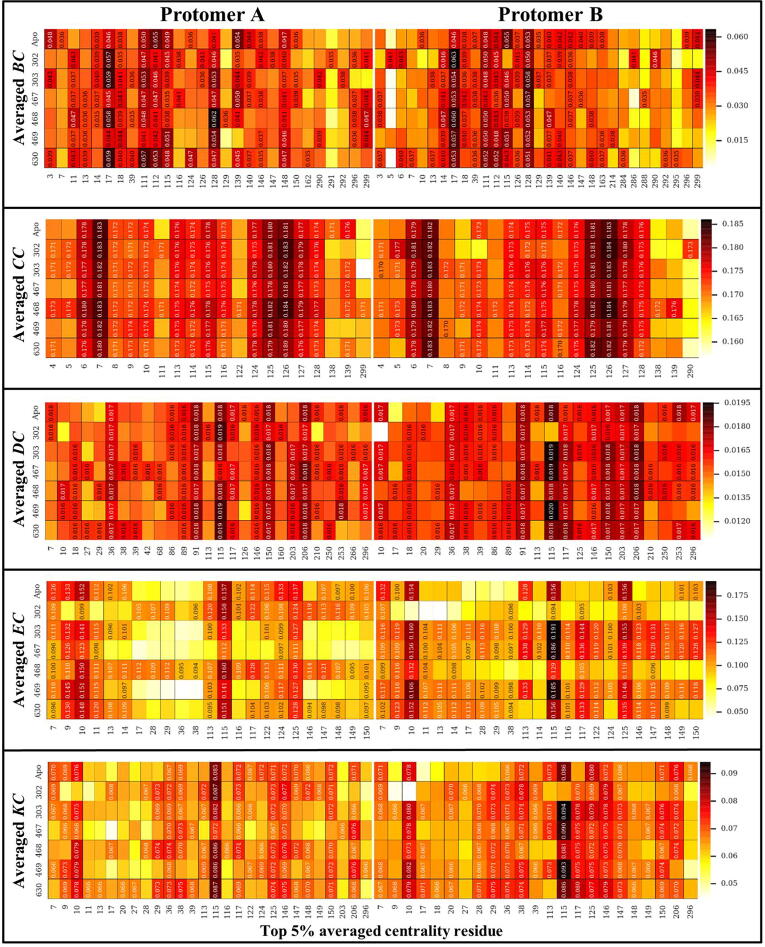
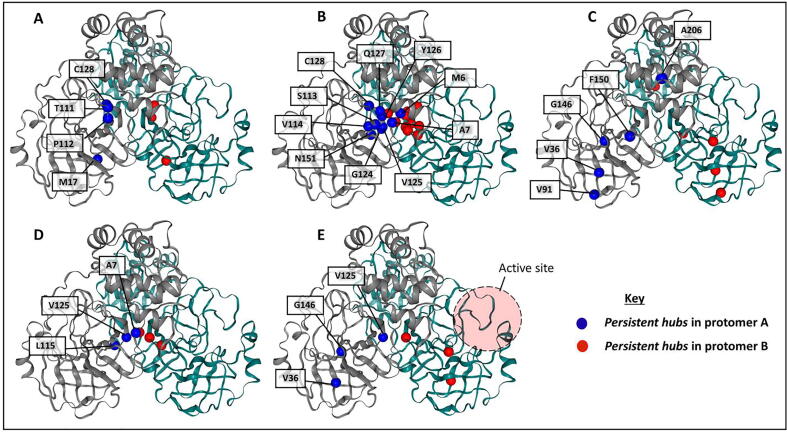
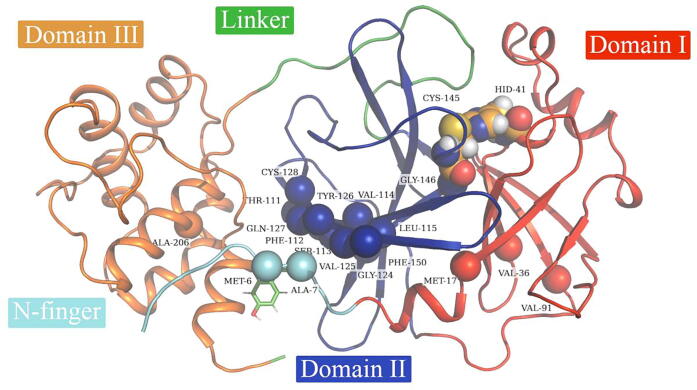
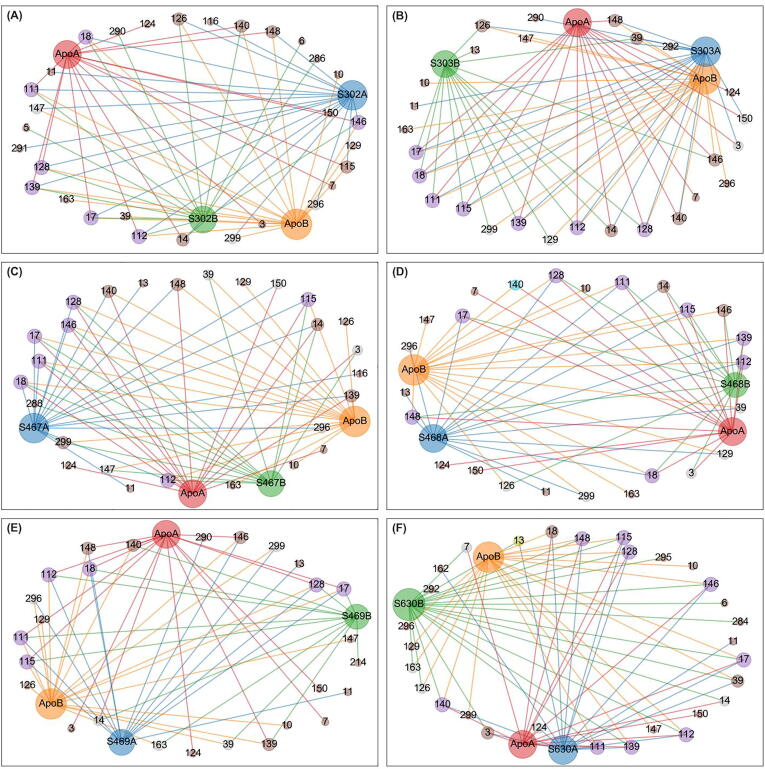
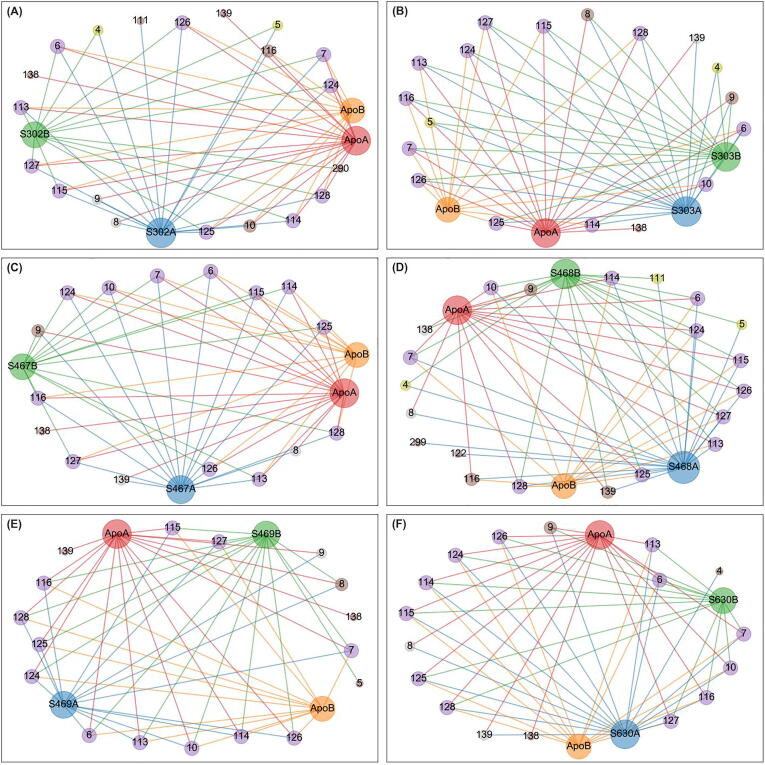
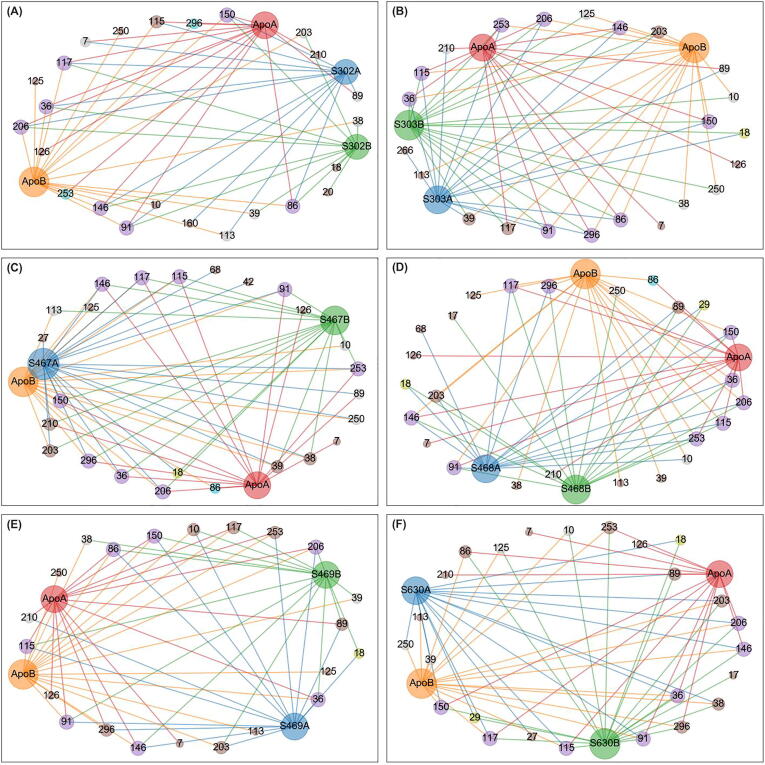
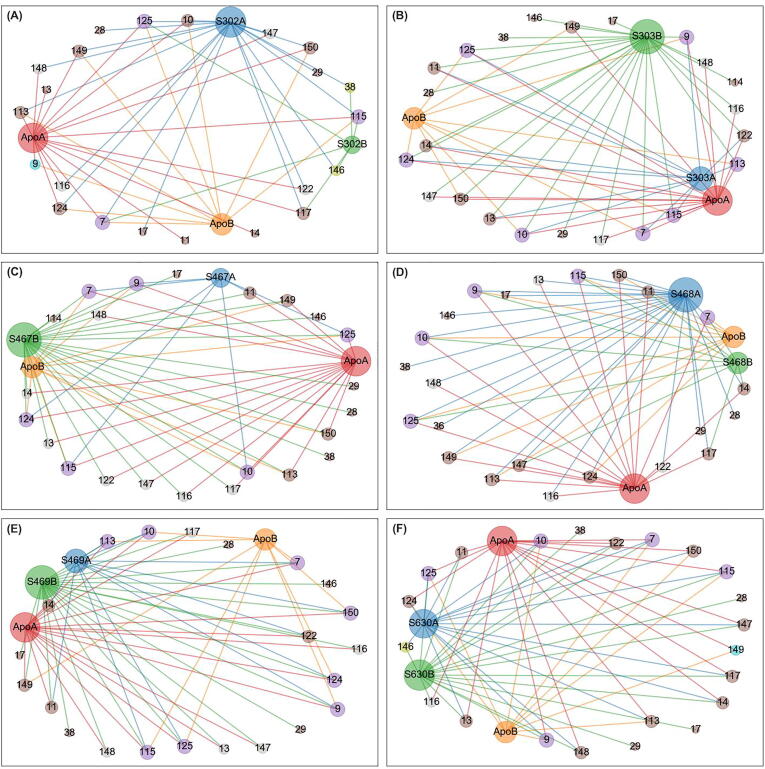
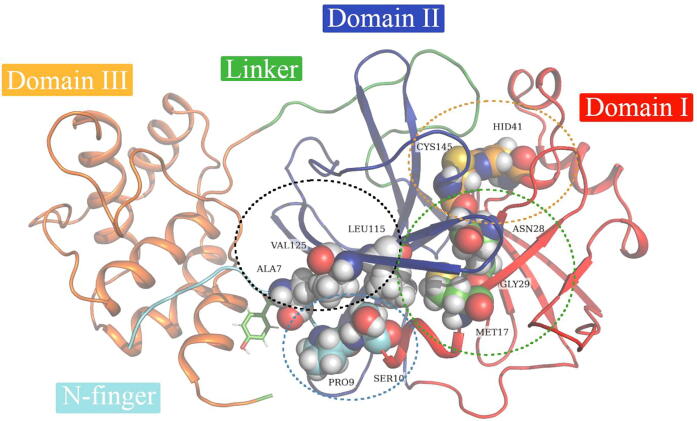
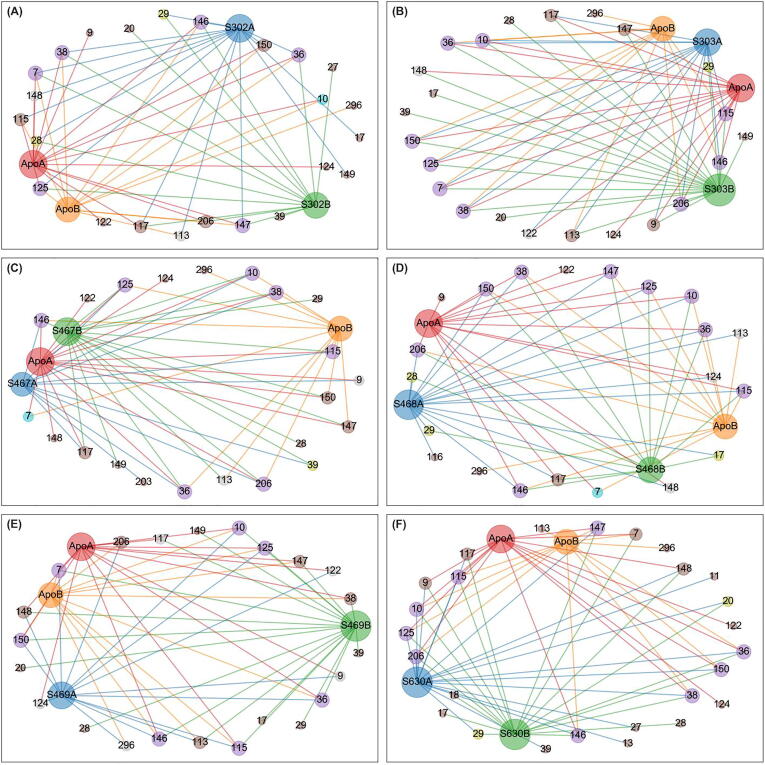
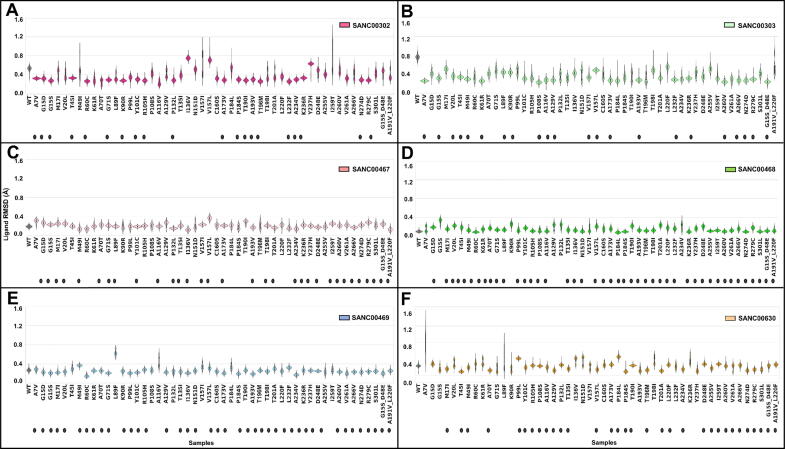
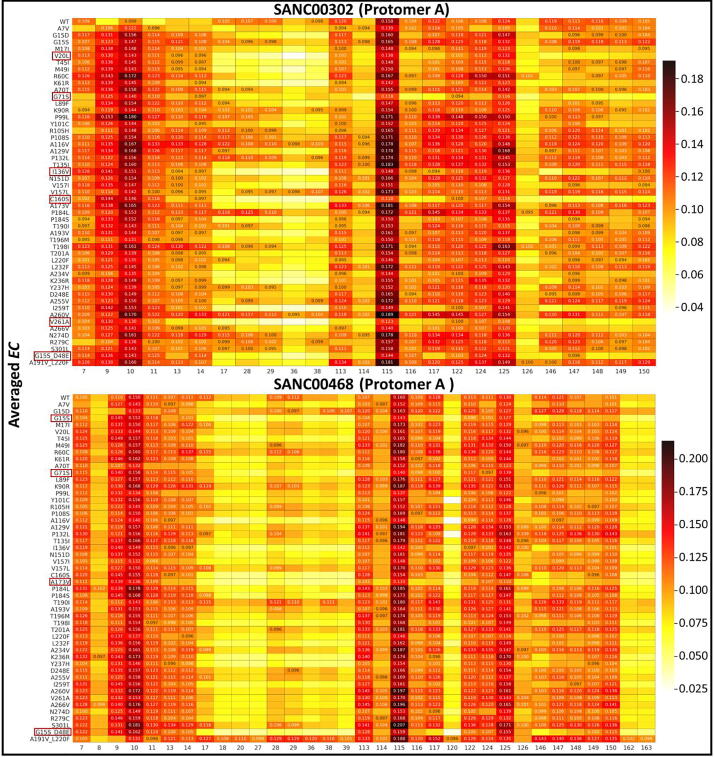
The rational search for allosteric modulators and the allosteric mechanisms of these modulators in the presence of mutations is a relatively unexplored field. Here, we established novel approaches and applied them to SARS-CoV-2 main protease (M) as a case study. First, we identified six potential allosteric modulators. Then, we focused on understanding the allosteric effects of these modulators on each of its protomers. We introduced a new combinatorial approach and dynamic residue network (DRN) analysis algorithms to examine patterns of change and conservation of critical nodes, according to five independent criteria of network centrality. We observed highly conserved network hubs for each averaged DRN metric on the basis of their existence in both protomers in the absence and presence of all ligands (). We also detected ligand specific signal changes. Using and ligand introduced hubs we identified a residue communication path connecting the allosteric binding site to the catalytic site. Finally, we examined the effects of the mutations on the behavior of the protein in the presence of selected potential allosteric modulators and investigated the ligand stability. One crucial outcome was to show that centrality hubs form an allosteric communication path between the allosteric ligand binding site to the active site going through the interface residues of domains I and II; and this path was either weakened or lost in the presence of some of the mutations. Overall, the results revealed crucial aspects that need to be considered in rational computational drug discovery.
在存在突变的情况下,对变构调节剂及其变构机制进行合理探索是一个相对未被充分研究的领域。在此,我们建立了新方法并将其应用于严重急性呼吸综合征冠状病毒2(SARS-CoV-2)主蛋白酶(M)作为案例研究。首先,我们鉴定出六种潜在的变构调节剂。然后,我们专注于了解这些调节剂对其每个原体的变构效应。我们引入了一种新的组合方法和动态残基网络(DRN)分析算法,根据网络中心性的五个独立标准来检查关键节点的变化和保守模式。基于在不存在和存在所有配体的情况下两个原体中均存在的情况,我们观察到每个平均DRN指标都有高度保守的网络枢纽。我们还检测到配体特异性信号变化。利用网络枢纽和引入配体的枢纽,我们确定了一条连接变构结合位点与催化位点的残基通信路径。最后,我们研究了突变对存在选定潜在变构调节剂时蛋白质行为的影响,并研究了配体稳定性。一个关键结果是表明中心性枢纽形成了一条从变构配体结合位点到活性位点的变构通信路径,该路径经过结构域I和II的界面残基;并且在存在某些突变的情况下,这条路径会减弱或消失。总体而言,结果揭示了合理计算药物发现中需要考虑的关键方面。