McMahon Aoife, Lewis Elizabeth, Buniello Annalisa, Cerezo Maria, Hall Peggy, Sollis Elliot, Parkinson Helen, Hindorff Lucia A, Harris Laura W, MacArthur Jacqueline A L
European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, UK.
Lead contact.
Cell Genom. 2021 Oct 13;1(1). doi: 10.1016/j.xgen.2021.100005.
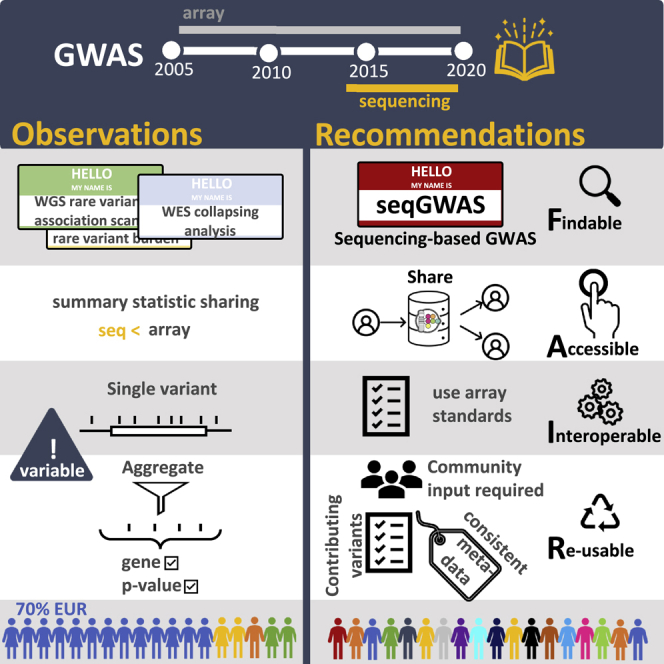
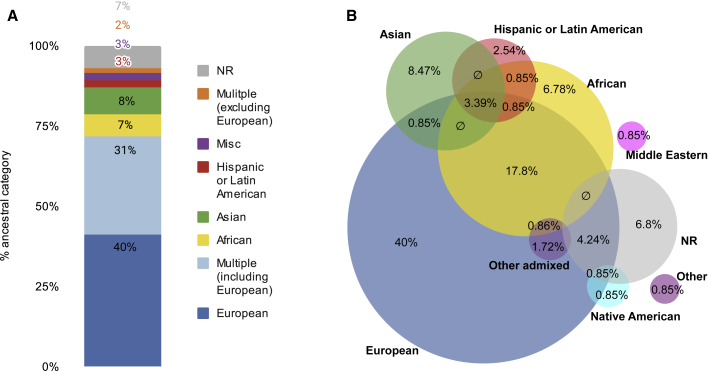
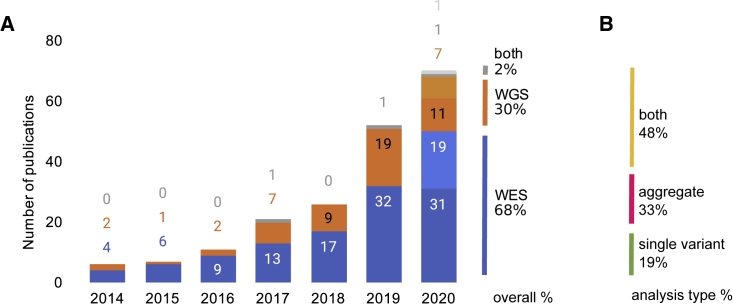
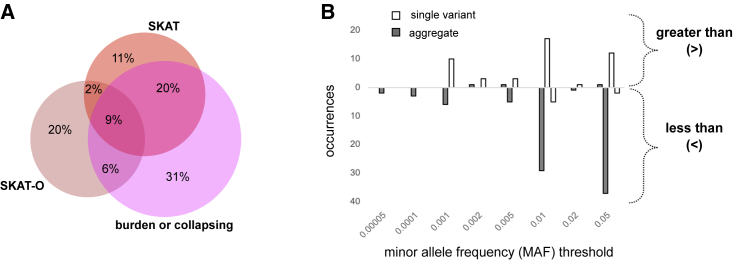
Genome sequencing has recently become a viable genotyping technology for use in genome-wide association studies (GWASs), offering the potential to analyze a broader range of genome-wide variation, including rare variants. To survey current standards, we assessed the content and quality of reporting of statistical methods, analyses, results, and datasets in 167 exome- or genome-wide-sequencing-based GWAS publications published from 2014 to 2020; 81% of publications included tests of aggregate association across multiple variants, with multiple test models frequently used. We observed a lack of standardized terms and incomplete reporting of datasets, particularly for variants analyzed in aggregate tests. We also find a lower frequency of sharing of summary statistics compared with array-based GWASs. Reporting standards and increased data sharing are required to ensure sequencing-based association study data are findable, interoperable, accessible, and reusable (FAIR). To support that, we recommend adopting the standard terminology of sequencing-based GWAS (seqGWAS). Further, we recommend that single-variant analyses be reported following the same standards and conventions as standard array-based GWASs and be shared in the GWAS Catalog. We also provide initial recommended standards for aggregate analyses metadata and summary statistics.
基因组测序最近已成为一种可行的基因分型技术,可用于全基因组关联研究(GWAS),有潜力分析更广泛的全基因组变异,包括罕见变异。为了审视当前标准,我们评估了2014年至2020年发表的167篇基于外显子组或全基因组测序的GWAS出版物中统计方法、分析、结果和数据集报告的内容与质量;81%的出版物包括对多个变异的总体关联测试,经常使用多种测试模型。我们发现缺乏标准化术语,且数据集报告不完整,尤其是在总体测试中分析的变异。我们还发现与基于芯片的GWAS相比,汇总统计数据的共享频率较低。需要报告标准和增加数据共享,以确保基于测序的关联研究数据是可查找、可互操作、可访问和可重复使用的(FAIR)。为了支持这一点,我们建议采用基于测序的GWAS(seqGWAS)的标准术语。此外,我们建议按照与基于标准芯片的GWAS相同的标准和惯例报告单变异分析,并在GWAS目录中共享。我们还提供了总体分析元数据和汇总统计数据的初步推荐标准。