Arastehfar A, Marcet-Houben M, Daneshnia F, Taj-Aldeen S J, Batra D, Lockhart S R, Shor E, Gabaldón T, Perlin D S
Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, NJ, 07110, USA.
Barcelona Supercomputing Centre (BSC-CNS), Jordi Girona 29, 08034, Barcelona, Spain.
Stud Mycol. 2021 Nov 29;100:100133. doi: 10.1016/j.simyco.2021.100133. eCollection 2021 Sep.
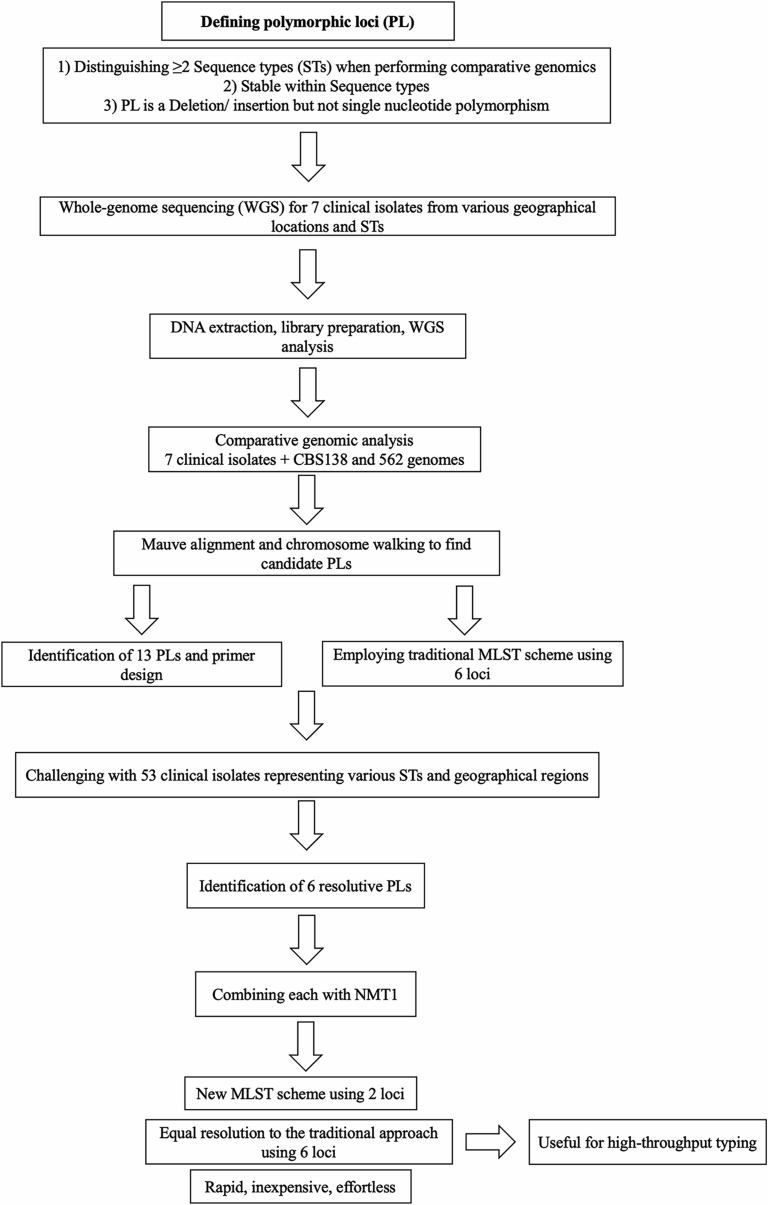
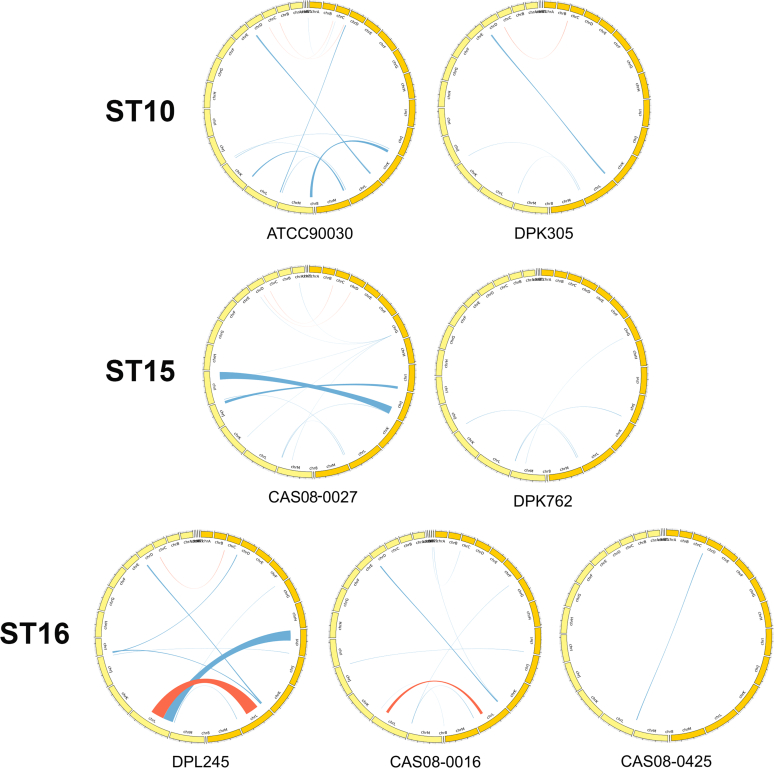
is the second leading cause of candidemia in many countries and is one of the most concerning yeast species of nosocomial importance due to its increasing rate of antifungal drug resistance and emerging multidrug-resistant isolates. Application of multilocus sequence typing (MLST) to clinical isolates revealed an association of certain sequence types (STs) with drug resistance and mortality. The current MLST scheme is based on single nucleotide polymorphisms (SNPs) at six loci and is therefore relatively laborious and costly. Furthermore, only a few high-quality reference genomes are available, limiting rapid analysis of clinical isolates by whole genome sequencing. In this study we provide long-read based assemblies for seven additional clinical strains belonging to three different STs and use this information to simplify the MLST scheme. Specifically, a comparison of these genomes identified highly polymorphic loci (HPL) defined by frequent insertions and deletions (indels), two of which proved to be highly resolutive for ST. When challenged with 53 additional isolates, a combination of (a component of the current MLST scheme) with either of the two HPL fully recapitulated ST identification. Therefore, our comparative genomic analysis identified a new typing approach combining SNPs and indels and based on only two loci, thus significantly simplifying ST identification in . Because typing tools are instrumental in addressing numerous clinical and biological questions, our new MLST scheme can be used for high throughput typing of in clinical and research settings.
在许多国家,它是念珠菌血症的第二大主要病因,并且由于其抗真菌药物耐药率不断上升以及出现多重耐药菌株,它是医院感染中最令人担忧的酵母菌种之一。将多位点序列分型(MLST)应用于临床分离株揭示了某些序列类型(STs)与耐药性和死亡率之间的关联。当前的MLST方案基于六个位点的单核苷酸多态性(SNP),因此相对费力且成本高昂。此外,仅有少数高质量的参考基因组可用,限制了通过全基因组测序对临床分离株进行快速分析。在本研究中,我们为属于三种不同STs的另外七个临床菌株提供了基于长读长的组装,并利用这些信息简化MLST方案。具体而言,对这些基因组的比较确定了由频繁插入和缺失(indels)定义的高度多态性位点(HPL),其中两个被证明对ST具有高度分辨力。当用另外53个分离株进行测试时,当前MLST方案的一个组成部分与两个HPL中的任何一个相结合,完全重现了ST鉴定。因此,我们的比较基因组分析确定了一种新的分型方法,该方法结合了SNP和indel且仅基于两个位点,从而显著简化了[该菌种名称]中的ST鉴定。由于分型工具对于解决众多临床和生物学问题至关重要,我们的新MLST方案可用于临床和研究环境中[该菌种名称]的高通量分型。