Ostner Johannes, Carcy Salomé, Müller Christian L
Department of Statistics, Ludwig-Maximilians-Universität München, Munich, Germany.
Institute of Computational Biology, Helmholtz Zentrum München, Munich, Germany.
Front Genet. 2021 Dec 7;12:766405. doi: 10.3389/fgene.2021.766405. eCollection 2021.
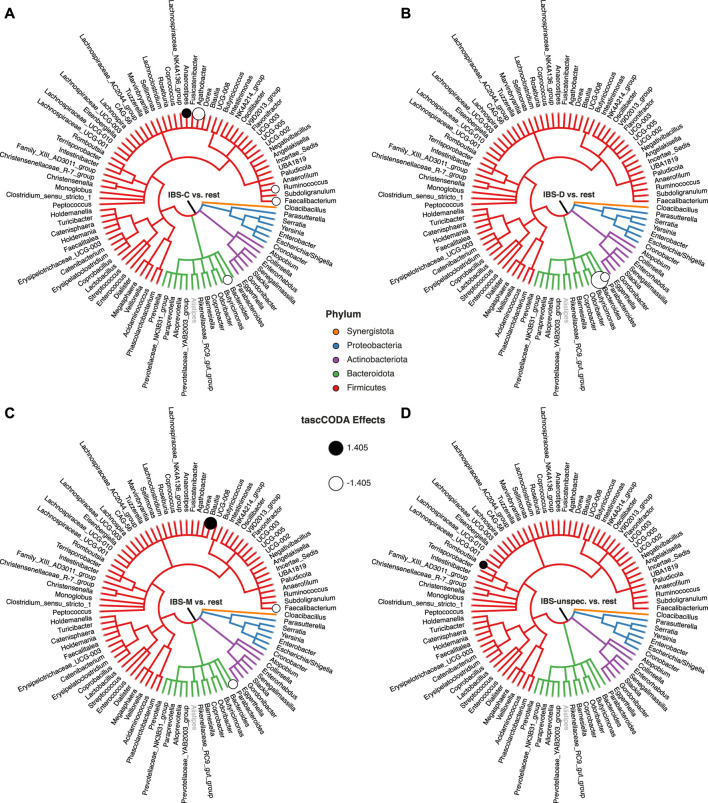
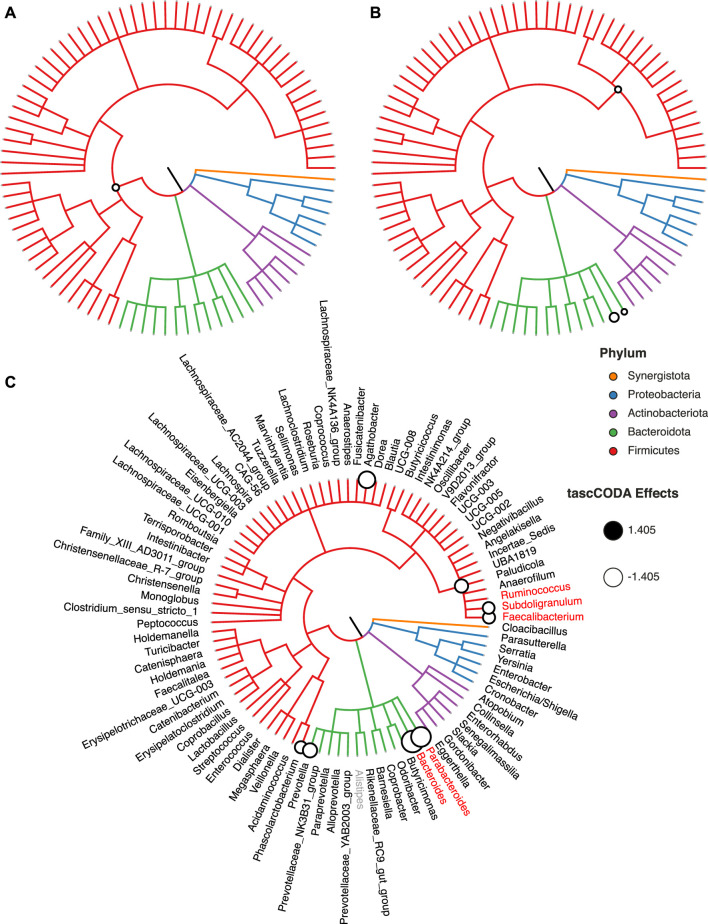
Accurate generative statistical modeling of count data is of critical relevance for the analysis of biological datasets from high-throughput sequencing technologies. Important instances include the modeling of microbiome compositions from amplicon sequencing surveys and the analysis of cell type compositions derived from single-cell RNA sequencing. Microbial and cell type abundance data share remarkably similar statistical features, including their inherent compositionality and a natural hierarchical ordering of the individual components from taxonomic or cell lineage tree information, respectively. To this end, we introduce a Bayesian model for ree-aggregated mplicon and ingle-ell mpositional ata nalysis (tascCODA) that seamlessly integrates hierarchical information and experimental covariate data into the generative modeling of compositional count data. By combining latent parameters based on the tree structure with spike-and-slab Lasso penalization, tascCODA can determine covariate effects across different levels of the population hierarchy in a data-driven parsimonious way. In the context of differential abundance testing, we validate tascCODA's excellent performance on a comprehensive set of synthetic benchmark scenarios. Our analyses on human single-cell RNA-seq data from ulcerative colitis patients and amplicon data from patients with irritable bowel syndrome, respectively, identified aggregated cell type and taxon compositional changes that were more predictive and parsimonious than those proposed by other schemes. We posit that tascCODA constitutes a valuable addition to the growing statistical toolbox for generative modeling and analysis of compositional changes in microbial or cell population data.
对计数数据进行准确的生成式统计建模对于高通量测序技术的生物数据集分析至关重要。重要的实例包括来自扩增子测序调查的微生物组组成建模以及源自单细胞RNA测序的细胞类型组成分析。微生物和细胞类型丰度数据具有非常相似的统计特征,分别包括其固有的组成性以及来自分类学或细胞谱系树信息的各个成分的自然层次排序。为此,我们引入了一种用于重新聚合扩增子和单细胞位置数据分析的贝叶斯模型(tascCODA),该模型将层次信息和实验协变量数据无缝集成到组成计数数据的生成建模中。通过将基于树结构的潜在参数与尖峰和平板Lasso惩罚相结合,tascCODA可以以数据驱动的简约方式确定不同人群层次水平上的协变量效应。在差异丰度测试的背景下,我们在一组全面的合成基准场景中验证了tascCODA的出色性能。我们分别对溃疡性结肠炎患者的人类单细胞RNA-seq数据和肠易激综合征患者的扩增子数据进行分析,确定了聚集的细胞类型和分类群组成变化,这些变化比其他方案提出的变化更具预测性和简约性。我们认为tascCODA是不断增长的用于生成建模和分析微生物或细胞群体数据组成变化的统计工具箱中的一个有价值的补充。