Family Medicine and Population Health (FAMPOP), Faculty of Medicine and Health Sciences, University of Antwerp, Wilrijk, Belgium.
FIND, Geneva, Switzerland.
Brief Bioinform. 2022 Jan 17;23(1). doi: 10.1093/bib/bbab541.
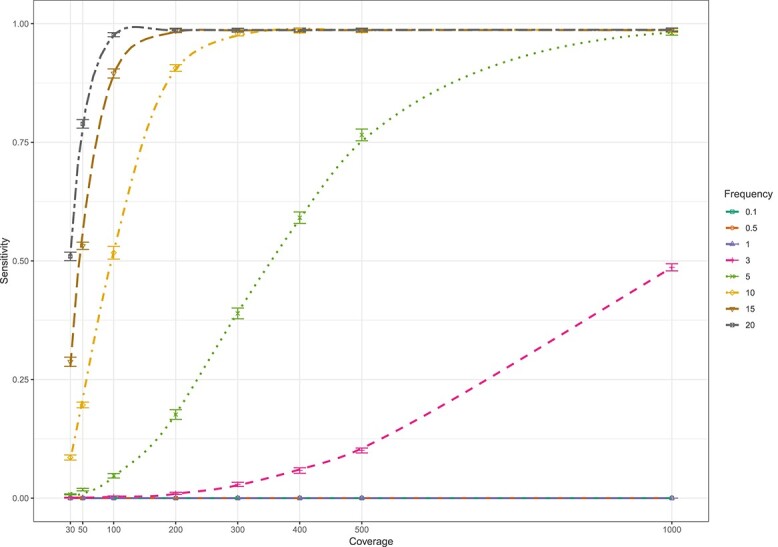
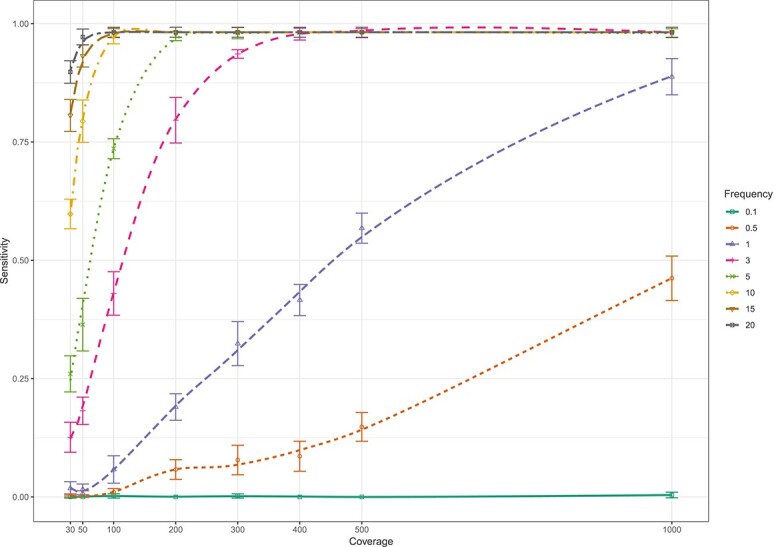
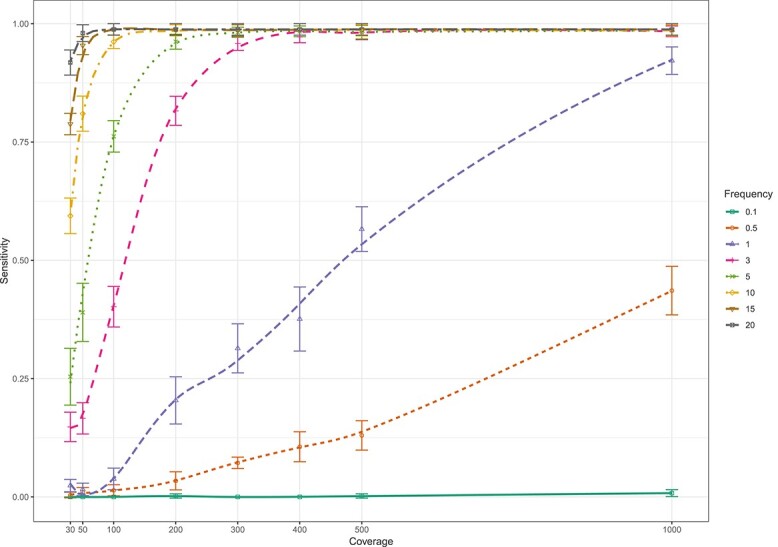
The study of genetic minority variants is fundamental to the understanding of complex processes such as evolution, fitness, transmission, virulence, heteroresistance and drug tolerance in Mycobacterium tuberculosis (Mtb). We evaluated the performance of the variant calling tool LoFreq to detect de novo as well as drug resistance conferring minor variants in both in silico and clinical Mtb next generation sequencing (NGS) data. The in silico simulations demonstrated that LoFreq is a conservative variant caller with very high precision (≥96.7%) over the entire range of depth of coverage tested (30x to1000x), independent of the type and frequency of the minor variant. Sensitivity increased with increasing depth of coverage and increasing frequency of the variant, and was higher for calling insertion and deletion (indel) variants than for single nucleotide polymorphisms (SNP). The variant frequency limit of detection was 0.5% and 3% for indel and SNP minor variants, respectively. For serial isolates from a patient with DR-TB; LoFreq successfully identified all minor Mtb variants in the Rv0678 gene (allele frequency as low as 3.22% according to targeted deep sequencing) in whole genome sequencing data (median coverage of 62X). In conclusion, LoFreq can successfully detect minor variant populations in Mtb NGS data, thus limiting the need for filtering of possible false positive variants due to sequencing error. The observed performance statistics can be used to determine the limit of detection in existing whole genome sequencing Mtb data and guide the required depth of future studies that aim to investigate the presence of minor variants.
研究遗传少数变体对于理解复杂过程至关重要,例如进化、适应性、传播、毒力、异质性耐药性和结核分枝杆菌(Mtb)的药物耐受性。我们评估了变体调用工具 LoFreq 在模拟和临床 Mtb 下一代测序(NGS)数据中检测从头变异以及赋予药物耐药性的少数变体的性能。模拟表明,LoFreq 是一种保守的变体调用器,在整个测试深度范围内(30x 到 1000x)具有非常高的精度(≥96.7%),与少数变体的类型和频率无关。随着深度的增加,灵敏度增加,而变体的频率增加,对插入和缺失(indel)变体的检测率高于单核苷酸多态性(SNP)。indel 和 SNP 少数变体的变体频率检测限分别为 0.5%和 3%。对于来自耐多药结核病(DR-TB)患者的连续分离株;LoFreq 成功地在全基因组测序数据(中位数覆盖度为 62X)中鉴定了 Rv0678 基因中的所有少数 Mtb 变体(根据靶向深度测序,等位基因频率低至 3.22%)。总之,LoFreq 可以成功地检测 Mtb NGS 数据中的少数变体群体,从而限制了由于测序错误而需要过滤可能的假阳性变体的需求。观察到的性能统计数据可用于确定现有全基因组测序 Mtb 数据中的检测限,并指导未来旨在研究少数变体存在性的研究所需的深度。