TORCH Consortium, Global Health Institute, Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium.
ADReM Data Lab, Department of Computer Science, University of Antwerp, Antwerp, Belgium.
PLoS Comput Biol. 2023 Nov 29;19(11):e1011648. doi: 10.1371/journal.pcbi.1011648. eCollection 2023 Nov.
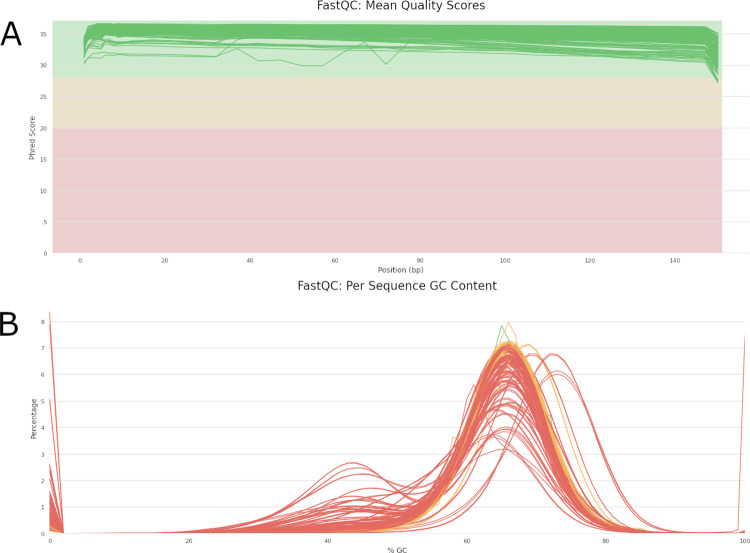
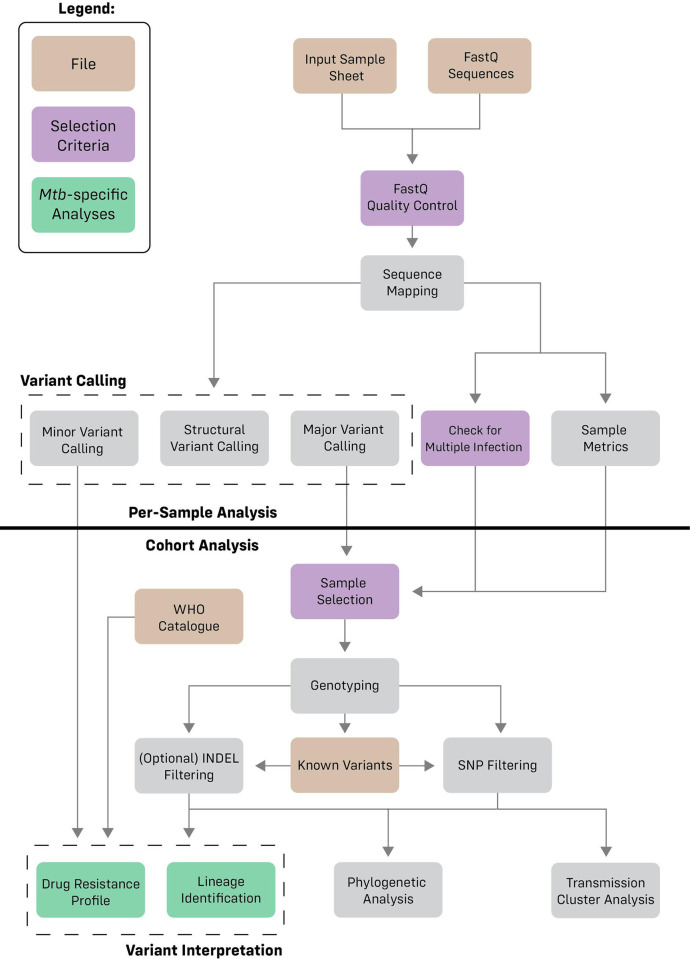
Whole genome sequencing (WGS) holds great potential for the management and control of tuberculosis. Accurate analysis of samples with low mycobacterial burden, which are characterized by low (<20x) coverage and high (>40%) levels of contamination, is challenging. We created the MAGMA (Maximum Accessible Genome for Mtb Analysis) bioinformatics pipeline for analysis of clinical Mtb samples.
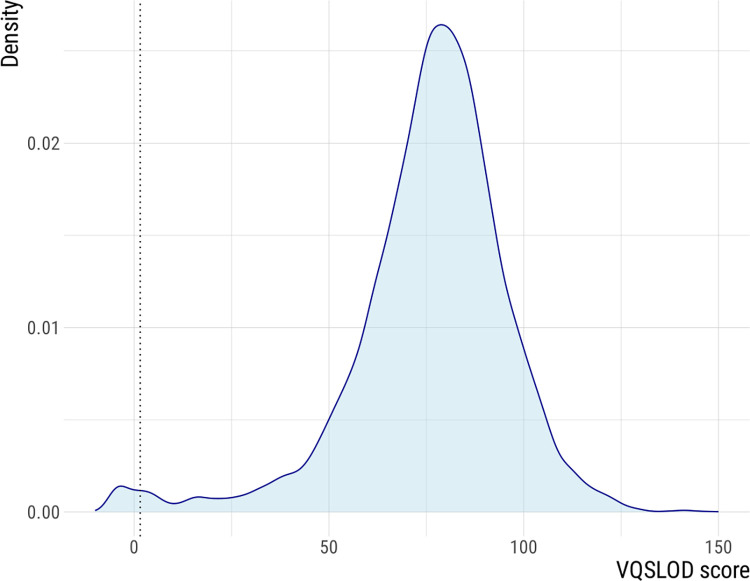
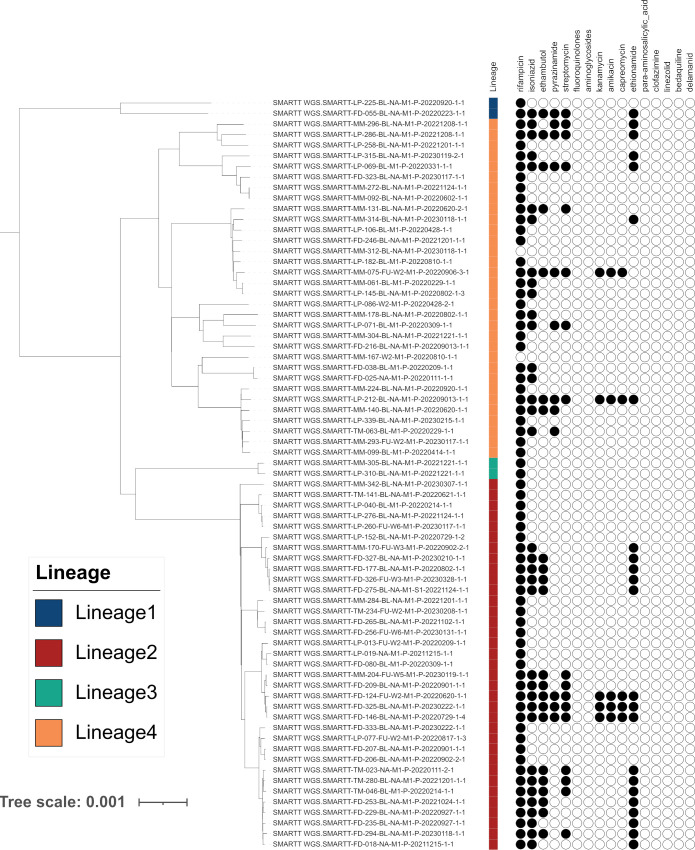
High accuracy variant calling is achieved by using a long seedlength during read mapping to filter out contaminants, variant quality score recalibration with machine learning to identify genuine genomic variants, and joint variant calling for low Mtb coverage genomes. MAGMA automatically generates a standardized and comprehensive output of drug resistance information and resistance classification based on the WHO catalogue of Mtb mutations. MAGMA automatically generates phylogenetic trees with drug resistance annotations and trees that visualize the presence of clusters. Drug resistance and phylogeny outputs from sequencing data of 79 primary liquid cultures were compared between the MAGMA and MTBseq pipelines. The MTBseq pipeline reported only a proportion of the variants in candidate drug resistance genes that were reported by MAGMA. Notable differences were in structural variants, variants in highly conserved rrs and rrl genes, and variants in candidate resistance genes for bedaquiline, clofazmine, and delamanid. Phylogeny results were similar between pipelines but only MAGMA visualized clusters.
The MAGMA pipeline could facilitate the integration of WGS into clinical care as it generates clinically relevant data on drug resistance and phylogeny in an automated, standardized, and reproducible manner.
全基因组测序(WGS)在结核病的管理和控制方面具有巨大潜力。分析低负荷分枝杆菌样本(覆盖度低(<20x)且污染度高(>40%))具有挑战性。我们创建了 MAGMA(用于 Mtb 分析的最大可访问基因组)生物信息学管道,用于分析临床 Mtb 样本。
通过在读取映射过程中使用长种子长度来过滤污染物,使用机器学习对变体质量评分进行重新校准以识别真正的基因组变体,以及对低 Mtb 覆盖基因组进行联合变体调用,实现了高精度的变体调用。MAGMA 会自动根据世界卫生组织的 Mtb 突变目录生成标准化和全面的耐药信息和耐药分类输出。MAGMA 会自动生成带有耐药注释和可视化聚类的进化树。对 79 个初始液体培养物测序数据的耐药性和进化树输出,在 MAGMA 和 MTBseq 管道之间进行了比较。MTBseq 管道仅报告了 MAGMA 报告的候选耐药基因中一部分变体。显著差异存在于结构变体、高度保守的 rrs 和 rrl 基因中的变体,以及候选耐药基因(贝达喹啉、氯法齐明和德拉马尼)中的变体。尽管两个管道的进化树结果相似,但只有 MAGMA 可视化了聚类。
MAGMA 管道可以促进 WGS 整合到临床护理中,因为它以自动化、标准化和可重复的方式生成关于耐药性和进化树的临床相关数据。