Division of Cancer Epidemiology and GeneticsDepartment of Health and Human Services, Medical Center Drive, National Cancer Institute, National Institutes of Health, Rockville, MD, USA.
Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA.
Breast Cancer Res. 2022 Jan 4;24(1):2. doi: 10.1186/s13058-021-01484-x.
Genome-wide association studies (GWAS) have identified multiple common breast cancer susceptibility variants. Many of these variants have differential associations by estrogen receptor (ER) status, but how these variants relate with other tumor features and intrinsic molecular subtypes is unclear.
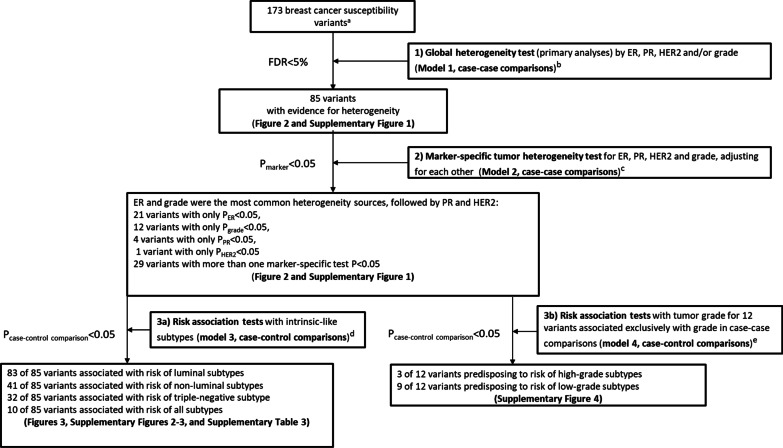
Among 106,571 invasive breast cancer cases and 95,762 controls of European ancestry with data on 173 breast cancer variants identified in previous GWAS, we used novel two-stage polytomous logistic regression models to evaluate variants in relation to multiple tumor features (ER, progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2) and grade) adjusting for each other, and to intrinsic-like subtypes.
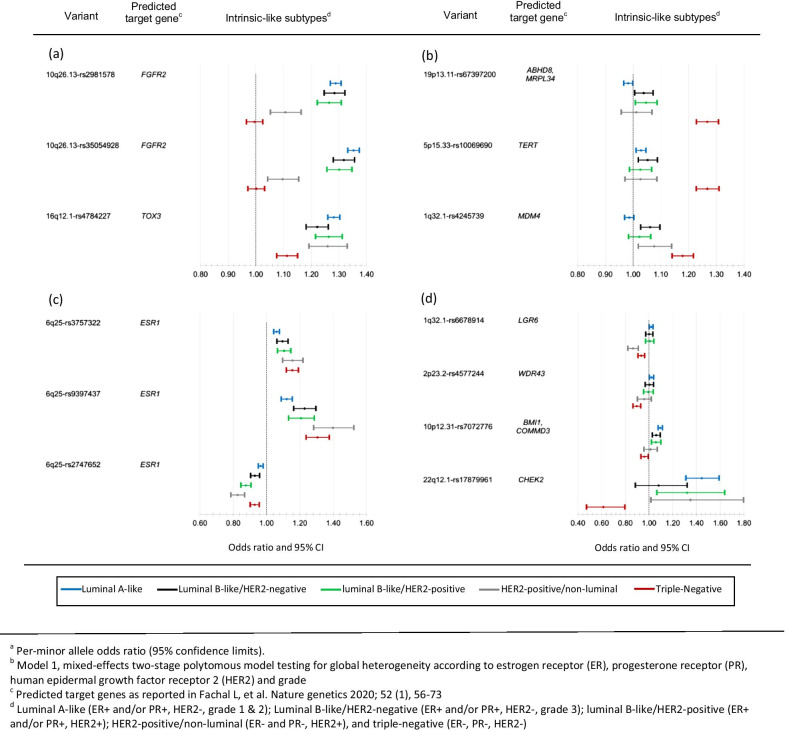
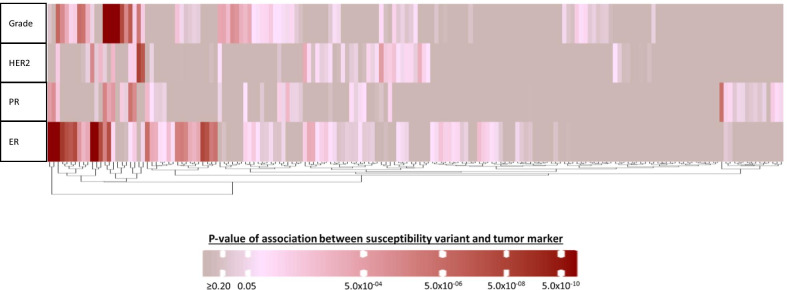
Eighty-five of 173 variants were associated with at least one tumor feature (false discovery rate < 5%), most commonly ER and grade, followed by PR and HER2. Models for intrinsic-like subtypes found nearly all of these variants (83 of 85) associated at p < 0.05 with risk for at least one luminal-like subtype, and approximately half (41 of 85) of the variants were associated with risk of at least one non-luminal subtype, including 32 variants associated with triple-negative (TN) disease. Ten variants were associated with risk of all subtypes in different magnitude. Five variants were associated with risk of luminal A-like and TN subtypes in opposite directions.
This report demonstrates a high level of complexity in the etiology heterogeneity of breast cancer susceptibility variants and can inform investigations of subtype-specific risk prediction.
全基因组关联研究(GWAS)已经确定了多个常见的乳腺癌易感性变异。这些变异中的许多与雌激素受体(ER)状态有关,但这些变异与其他肿瘤特征和内在分子亚型的关系尚不清楚。
在 106571 例浸润性乳腺癌病例和 95762 例欧洲裔对照者中,我们使用新的两阶段多分类逻辑回归模型,评估了与多种肿瘤特征(ER、孕激素受体(PR)、人表皮生长因子受体 2(HER2)和分级)相关的 173 个先前 GWAS 中发现的乳腺癌变异,这些特征相互调整,并与内在相似的亚型相关。
在 173 个变异中,有 85 个与至少一个肿瘤特征相关(假发现率<5%),最常见的是 ER 和分级,其次是 PR 和 HER2。内在相似亚型的模型发现,几乎所有这些变异(85 个中的 83 个)都与至少一种 luminal 样亚型的风险相关(p<0.05),大约一半(85 个中的 41 个)的变异与至少一种非 luminal 亚型的风险相关,包括 32 个与三阴性(TN)疾病相关的变异。有 10 个变异与所有亚型的风险相关,程度不同。有 5 个变异与 luminal A 样和 TN 亚型的风险呈相反方向相关。
本报告表明乳腺癌易感性变异的病因异质性程度很高,可为亚型特异性风险预测的研究提供信息。