Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY, 10029, USA.
Mount Sinai Center for Transformative Disease Modeling, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY, 10029, USA.
Mol Neurodegener. 2022 Jan 9;17(1):5. doi: 10.1186/s13024-021-00507-7.
Cellular senescence is a complex stress response that impacts cellular function and organismal health. Multiple developmental and environmental factors, such as intrinsic cellular cues, radiation, oxidative stress, oncogenes, and protein accumulation, activate genes and pathways that can lead to senescence. Enormous efforts have been made to identify and characterize senescence genes (SnGs) in stress and disease systems. However, the prevalence of senescent cells in healthy human tissues and the global SnG expression signature in different cell types are poorly understood.
This study performed an integrative gene network analysis of bulk and single-cell RNA-seq data in non-diseased human tissues to investigate SnG co-expression signatures and their cell-type specificity.
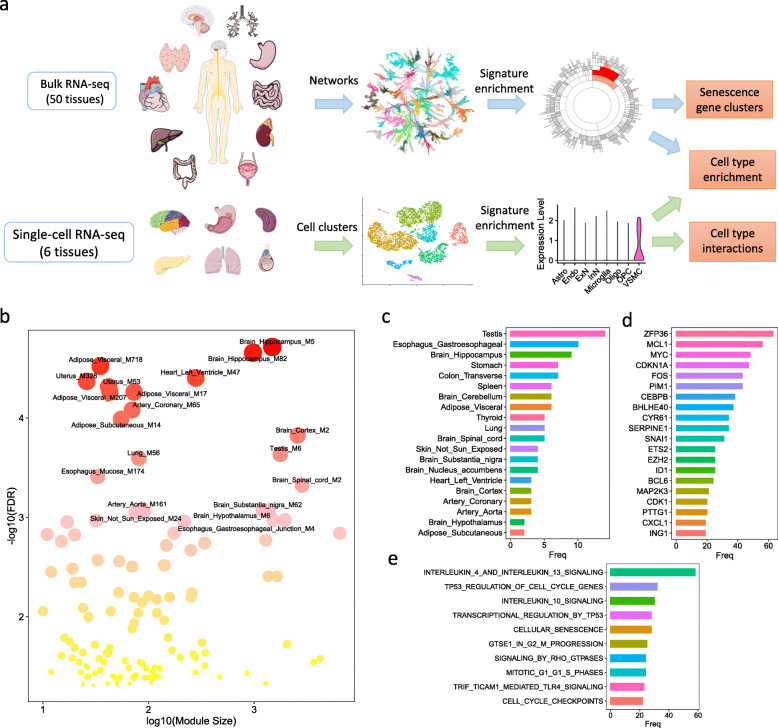
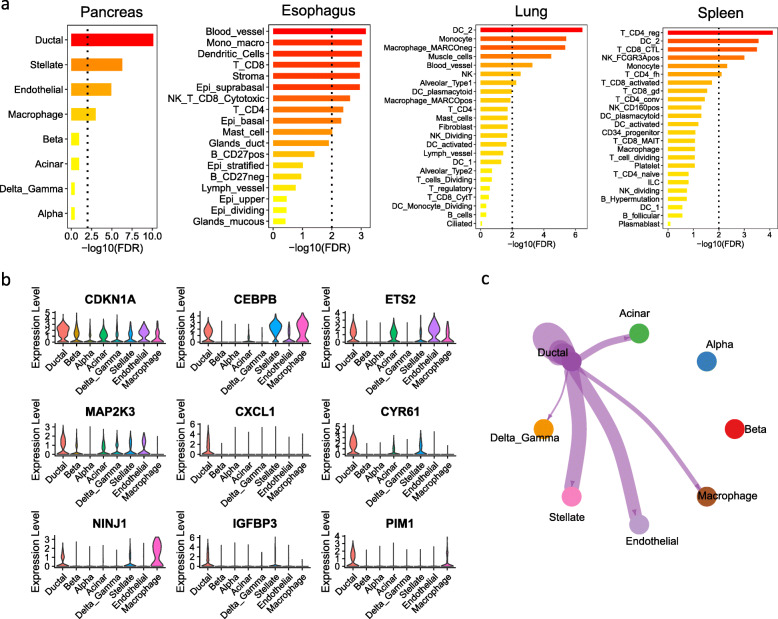
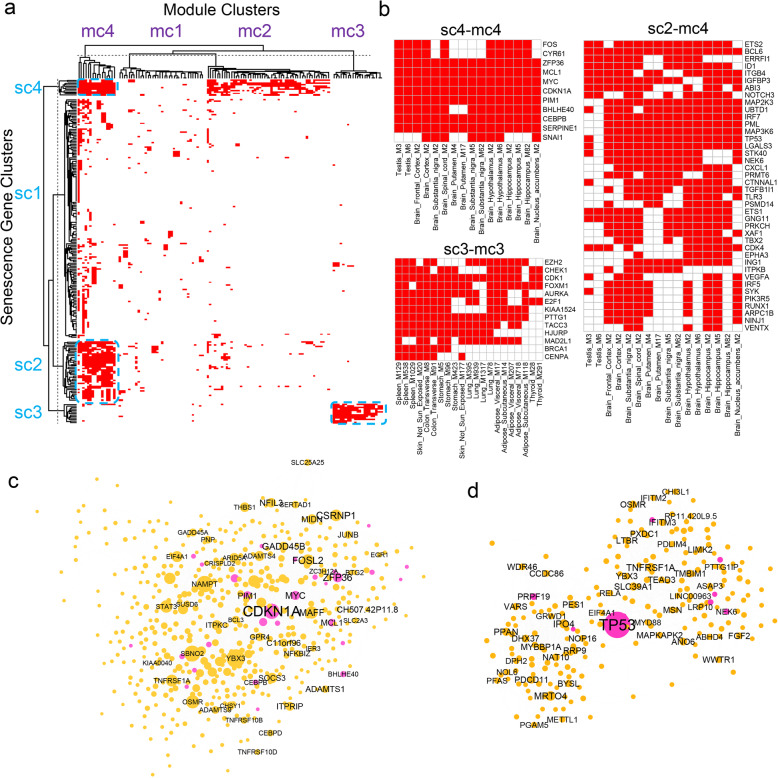
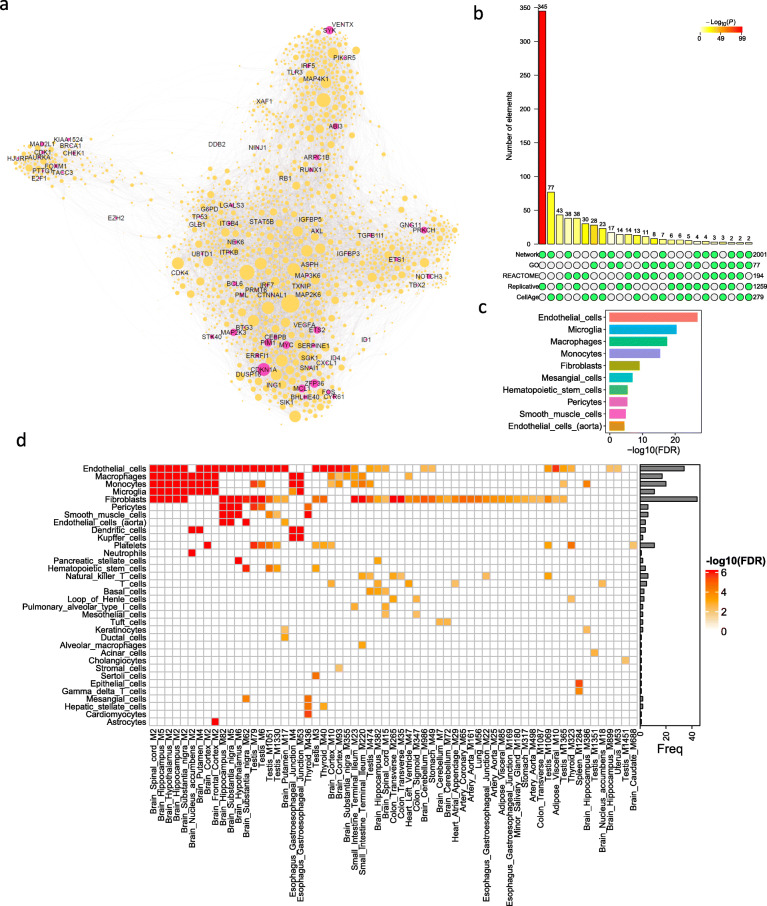
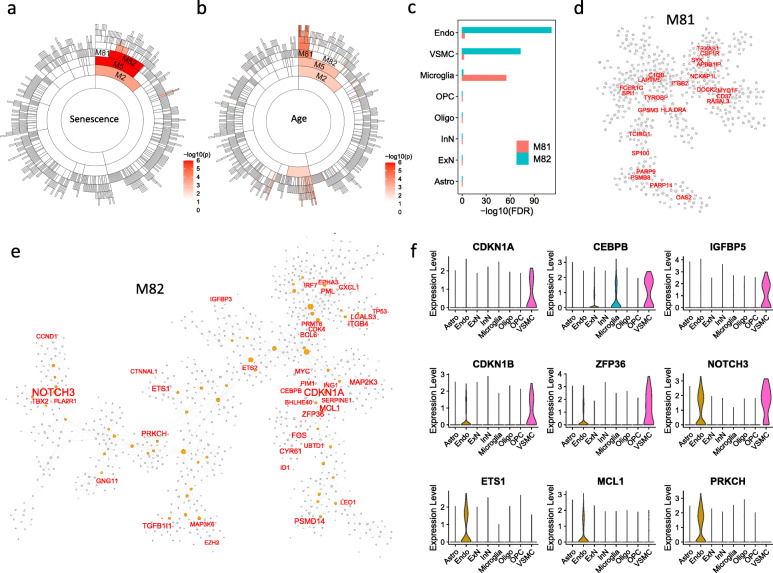
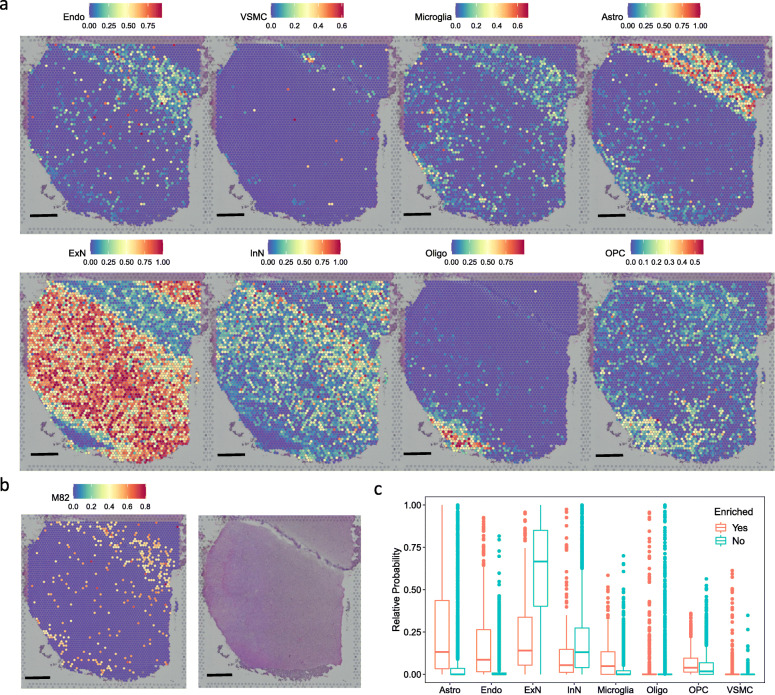
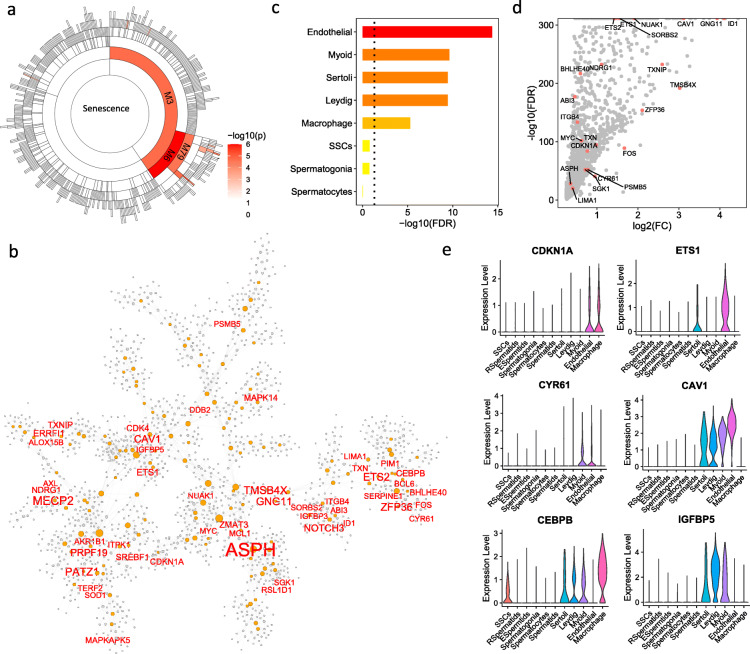
Through a comprehensive transcriptomic network analysis of 50 human tissues in the Genotype-Tissue Expression Project (GTEx) cohort, we identified SnG-enriched gene modules, characterized SnG co-expression patterns, and constructed aggregated SnG networks across primary tissues of the human body. Our network approaches identified 51 SnGs highly conserved across the human tissues, including CDKN1A (p21)-centered regulators that control cell cycle progression and the senescence-associated secretory phenotype (SASP). The SnG-enriched modules showed remarkable cell-type specificity, especially in fibroblasts, endothelial cells, and immune cells. Further analyses of single-cell RNA-seq and spatial transcriptomic data independently validated the cell-type specific SnG signatures predicted by the network analysis.
This study systematically revealed the co-regulated organizations and cell type specificity of SnGs in major human tissues, which can serve as a blueprint for future studies to map senescent cells and their cellular interactions in human tissues.
细胞衰老(cellular senescence)是一种复杂的应激反应,会影响细胞功能和机体健康。多种发育和环境因素,如内在的细胞信号、辐射、氧化应激、致癌基因和蛋白质积累,会激活导致衰老的基因和途径。人们已经做出了巨大的努力来识别和描述应激和疾病系统中的衰老相关基因(SnGs)。然而,健康人体组织中衰老细胞的普遍性以及不同细胞类型中的全局 SnG 表达特征仍知之甚少。
本研究对非疾病人类组织中的批量和单细胞 RNA-seq 数据进行了综合基因网络分析,以研究 SnG 的共表达特征及其细胞类型特异性。
通过对基因型组织表达项目(GTEx)队列中 50 个人类组织的综合转录组网络分析,我们确定了 SnG 富集的基因模块,描述了 SnG 的共表达模式,并构建了人体主要组织的综合 SnG 网络。我们的网络方法确定了 51 个在人类组织中高度保守的 SnGs,包括控制细胞周期进程和衰老相关分泌表型(SASP)的 CDKN1A(p21)为中心的调节因子。SnG 富集的模块表现出显著的细胞类型特异性,尤其是在成纤维细胞、内皮细胞和免疫细胞中。对单细胞 RNA-seq 和空间转录组数据的进一步分析独立验证了网络分析预测的细胞类型特异性 SnG 特征。
本研究系统地揭示了主要人类组织中 SnGs 的共调控组织和细胞类型特异性,可为未来研究绘制人类组织中衰老细胞及其细胞相互作用提供蓝图。