Chen Xiaomin, Wang Ruoyu, Xu Tianze, Zhang Yajing, Li Hongyan, Du Chengcheng, Wang Kun, Gao Zairong
Department of Nuclear Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Hubei Province Key Laboratory of Molecular Imaging, Wuhan, China.
Transl Cancer Res. 2021 Feb;10(2):694-713. doi: 10.21037/tcr-20-2866.
The genes and genetic mechanisms underlying the occurrence and progression of papillary thyroid carcinoma (PTC) are still unknown. This study aimed to find candidate genes related to the pathogenesis and progression of PTC.
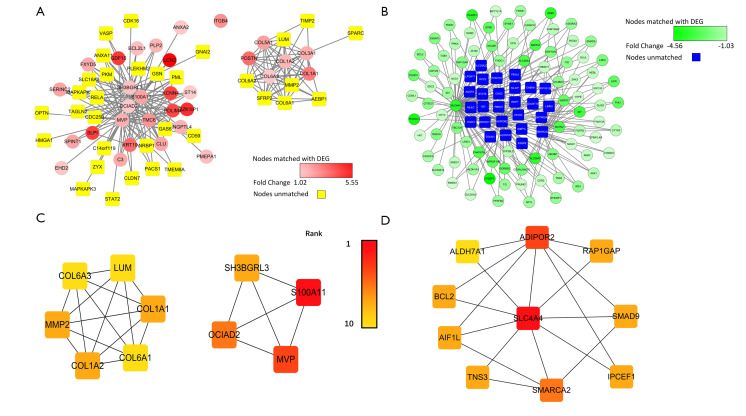
RNA sequencing (RNA-seq) data of PTC and normal tissues were downloaded from The Cancer Genome Atlas (TCGA) database with clinical stage data to form a test, validation, and clinical-stage data matrix. We used the test data set to analyze differentially expressed genes (DEGs) and weighted gene co-expression network analysis (WGCNA) to find those gene clusters highly correlated with PTC. We then verified the expression of genes in the interested modules using the validation matrix. The quantitative real-time polymerase chain reaction (qRT-PCR) was used to verify the reliability of the expression of selected genes. Five key genes (, , , , and ) were used to analyze the connection between gene expression and the American Joint Committee on Cancer (AJCC) stage. The upregulated and downregulated DEGs, along with the modules of interest, were subjected to Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment using the Database for Annotation, Visualization, and Integrated Discovery (DAVID).
We used WGCNA to find two modules of interest, the yellow module, which was positively associated with PTC, and the blue module, which was negatively correlated with PTC. Four genes (, , , and ) from the yellow module were determined to be highly expressed in PTC in the test data matrix and were verified in both the validation data matrix and quantitative real-time PCR, which indicated that these four genes were highly correlated with the occurrence of the PTC. Furthermore, these four genes also had a significantly higher expression in the advanced levels of pathological T, N, and AJCC stage, meaning that they were correlated with the progression of PTC. Genes in the yellow module and upregulated DEGs were significantly enriched in three vital signaling pathways, including focal adhesion, extracellular matrix (ECM)-receptor interaction, and the PI3K-Akt signaling pathway.
Four candidate genes (, , , and ) may be potential biomarkers for the PTC's pathogenesis and may be useful for predicting the disease stage.
甲状腺乳头状癌(PTC)发生和进展的基因及遗传机制仍不清楚。本研究旨在寻找与PTC发病机制和进展相关的候选基因。
从癌症基因组图谱(TCGA)数据库下载PTC和正常组织的RNA测序(RNA-seq)数据以及临床分期数据,形成测试、验证和临床分期数据矩阵。我们使用测试数据集分析差异表达基因(DEG),并通过加权基因共表达网络分析(WGCNA)来寻找与PTC高度相关的基因簇。然后我们使用验证矩阵验证感兴趣模块中基因的表达。采用定量实时聚合酶链反应(qRT-PCR)验证所选基因表达的可靠性。使用五个关键基因(、、、和)分析基因表达与美国癌症联合委员会(AJCC)分期之间的联系。上调和下调的DEG以及感兴趣的模块,使用注释、可视化和综合发现数据库(DAVID)进行基因本体论(GO)富集和京都基因与基因组百科全书(KEGG)通路富集。
我们使用WGCNA发现了两个感兴趣的模块,黄色模块与PTC呈正相关,蓝色模块与PTC呈负相关。在测试数据矩阵中,确定黄色模块中的四个基因(、、、和)在PTC中高表达,并在验证数据矩阵和定量实时PCR中得到验证,这表明这四个基因与PTC的发生高度相关。此外,这四个基因在病理T、N和AJCC分期的晚期水平也有显著更高的表达,这意味着它们与PTC的进展相关。黄色模块中的基因和上调的DEG在三个重要信号通路中显著富集,包括粘着斑、细胞外基质(ECM)-受体相互作用和PI3K-Akt信号通路。
四个候选基因(、、、和)可能是PTC发病机制的潜在生物标志物,可能有助于预测疾病分期。