Molecular and Experimental Mycobacteriology, Research Center Borstel, Borstel, Germany.
German Center for Infection Research (DZIF), Partner Site Hamburg-Lübeck-Borstel-Riems, Hamburg, Germany.
Genome Med. 2022 Feb 9;14(1):13. doi: 10.1186/s13073-022-01017-x.
Bacteria belonging to the genus Haemophilus cause a wide range of diseases in humans. Recently, H. influenzae was classified by the WHO as priority pathogen due to the wide spread of ampicillin resistant strains. However, other Haemophilus spp. are often misclassified as H. influenzae. Therefore, we established an accurate and rapid whole genome sequencing (WGS) based classification and serotyping algorithm and combined it with the detection of resistance genes.
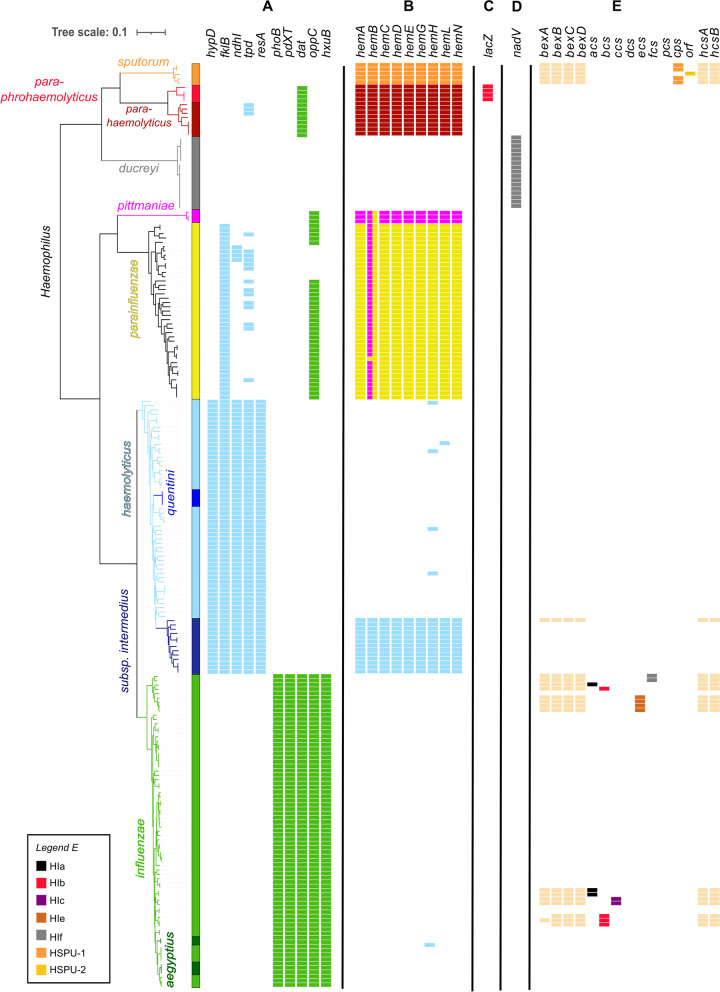
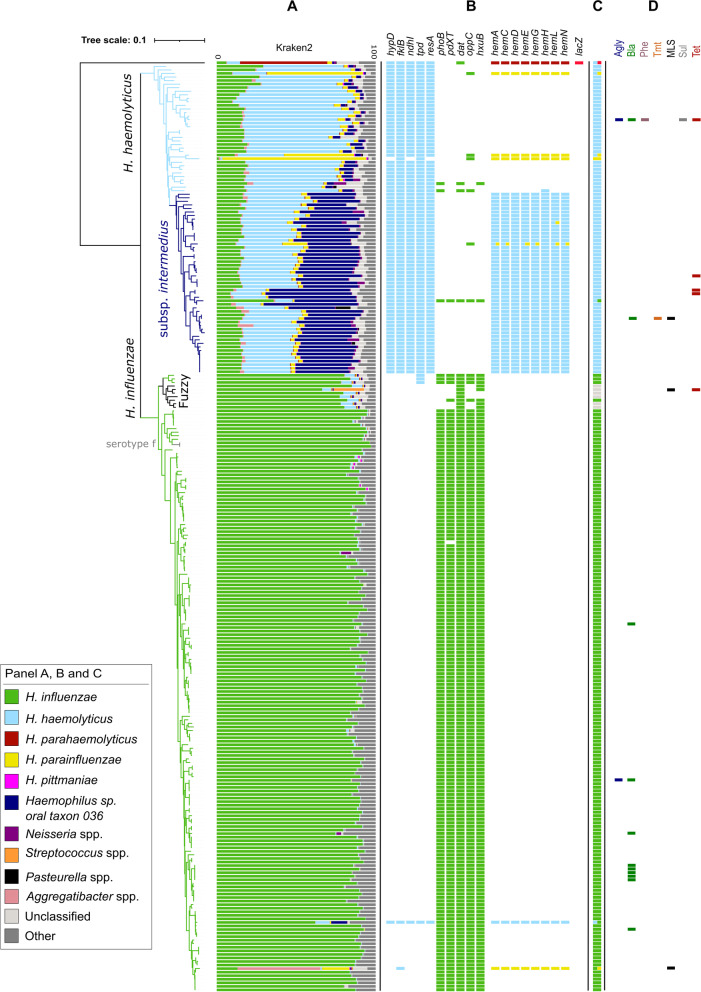
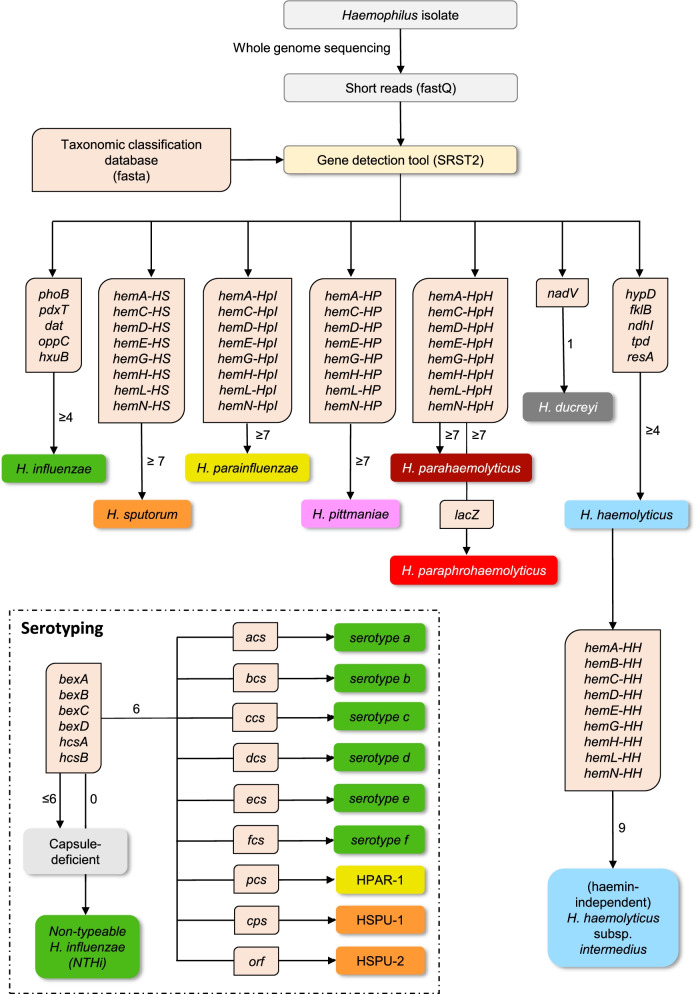
A gene presence/absence-based classification algorithm was developed, which employs the open-source gene-detection tool SRST2 and a new classification database comprising 36 genes, including capsule loci for serotyping. These genes were identified using a comparative genome analysis of 215 strains belonging to ten human-related Haemophilus (sub)species (training dataset). The algorithm was evaluated on 1329 public short read datasets (evaluation dataset) and used to reclassify 262 clinical Haemophilus spp. isolates from 250 patients (German cohort). In addition, the presence of antibiotic resistance genes within the German dataset was evaluated with SRST2 and correlated with results of traditional phenotyping assays.
The newly developed algorithm can differentiate between clinically relevant Haemophilus species including, but not limited to, H. influenzae, H. haemolyticus, and H. parainfluenzae. It can also identify putative haemin-independent H. haemolyticus strains and determine the serotype of typeable Haemophilus strains. The algorithm performed excellently in the evaluation dataset (99.6% concordance with reported species classification and 99.5% with reported serotype) and revealed several misclassifications. Additionally, 83 out of 262 (31.7%) suspected H. influenzae strains from the German cohort were in fact H. haemolyticus strains, some of which associated with mouth abscesses and lower respiratory tract infections. Resistance genes were detected in 16 out of 262 datasets from the German cohort. Prediction of ampicillin resistance, associated with bla, and tetracycline resistance, associated with tetB, correlated well with available phenotypic data.
Our new classification database and algorithm have the potential to improve diagnosis and surveillance of Haemophilus spp. and can easily be coupled with other public genotyping and antimicrobial resistance databases. Our data also point towards a possible pathogenic role of H. haemolyticus strains, which needs to be further investigated.
嗜血杆菌属的细菌可引起人类多种疾病。最近,世卫组织将流感嗜血杆菌列为优先病原体,因为氨苄西林耐药菌株广泛传播。然而,其他嗜血杆菌属通常被错误分类为流感嗜血杆菌。因此,我们建立了一种准确、快速的全基因组测序(WGS)分类和血清分型算法,并将其与耐药基因检测相结合。
开发了一种基于基因存在/缺失的分类算法,该算法采用开源基因检测工具 SRST2 和一个新的分类数据库,该数据库包含 36 个基因,包括用于血清分型的荚膜基因座。这些基因是通过对 215 株与人相关的嗜血杆菌(亚)种(训练数据集)的比较基因组分析确定的。该算法在 1329 个公共短读数据集(评估数据集)上进行了评估,并用于重新分类 250 名患者的 262 株临床嗜血杆菌分离株(德国队列)。此外,使用 SRST2 评估德国数据集内抗生素耐药基因的存在,并将其与传统表型分析结果相关联。
新开发的算法可区分临床相关的嗜血杆菌种,包括但不限于流感嗜血杆菌、溶血嗜血杆菌和副流感嗜血杆菌。它还可以识别潜在的血红素非依赖型溶血嗜血杆菌菌株,并确定可分型嗜血杆菌菌株的血清型。该算法在评估数据集(与报告的物种分类的一致性为 99.6%,与报告的血清型的一致性为 99.5%)中表现出色,并揭示了一些错误分类。此外,德国队列中 262 株疑似流感嗜血杆菌分离株中有 83 株(31.7%)实际上是溶血嗜血杆菌菌株,其中一些与口腔脓肿和下呼吸道感染有关。在德国队列的 262 个数据集的 16 个中检测到耐药基因。与 bla 相关的氨苄西林耐药和与 tetB 相关的四环素耐药的预测与可用的表型数据相关性良好。
我们的新分类数据库和算法有可能改善嗜血杆菌属的诊断和监测,并且可以很容易地与其他公共基因分型和抗菌药物耐药性数据库相结合。我们的数据还表明溶血嗜血杆菌菌株可能具有致病性,这需要进一步研究。