Xia Yu, Zhao Yu-Dong, Sun Gui-Xiang, Xia Shuai-Shuai, Yang Zheng-Wang
Provincial Key Laboratory of TCM Diagnostics, Hunan University of Chinese Medicine, Changsha, Hunan Province, 410208, People's Republic of China.
Institute of Chinese Medicine Diagnosis, Hunan University of Chinese Medicine, Changsha, Hunan Province, 410208, People's Republic of China.
Int J Gen Med. 2022 Feb 2;15:1023-1032. doi: 10.2147/IJGM.S348175. eCollection 2022.
Preeclampsia (PE) is a pregnancy-specific multisystem disease as well as an important cause of maternal and perinatal death. This study aimed to analyze the placental transcriptional data and clinical information of PE patients available in the published database and predict the target genes for prevention of PE.
The clinical information and corresponding RNA data of PE patients were downloaded from the GEO database. Cluster analysis was performed to examine the correlation between different genotyping genes and clinical manifestations. Then, bioinformatic approaches including GO, KEGG, WGCNA, and GSEA were employed to functionally characterize candidate target genes involved in pathogenesis of PE.
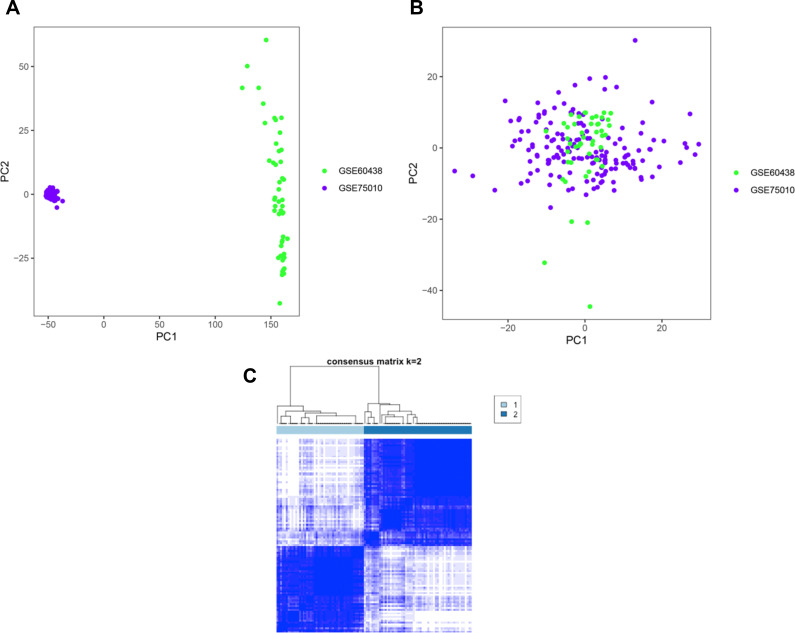
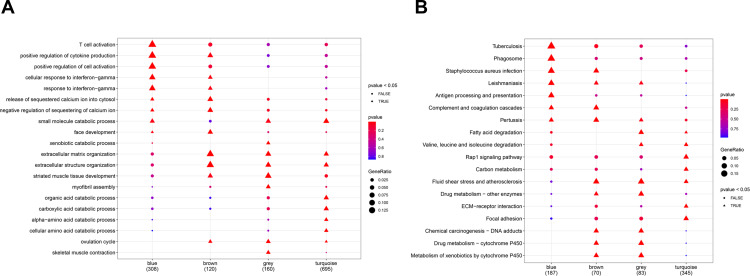
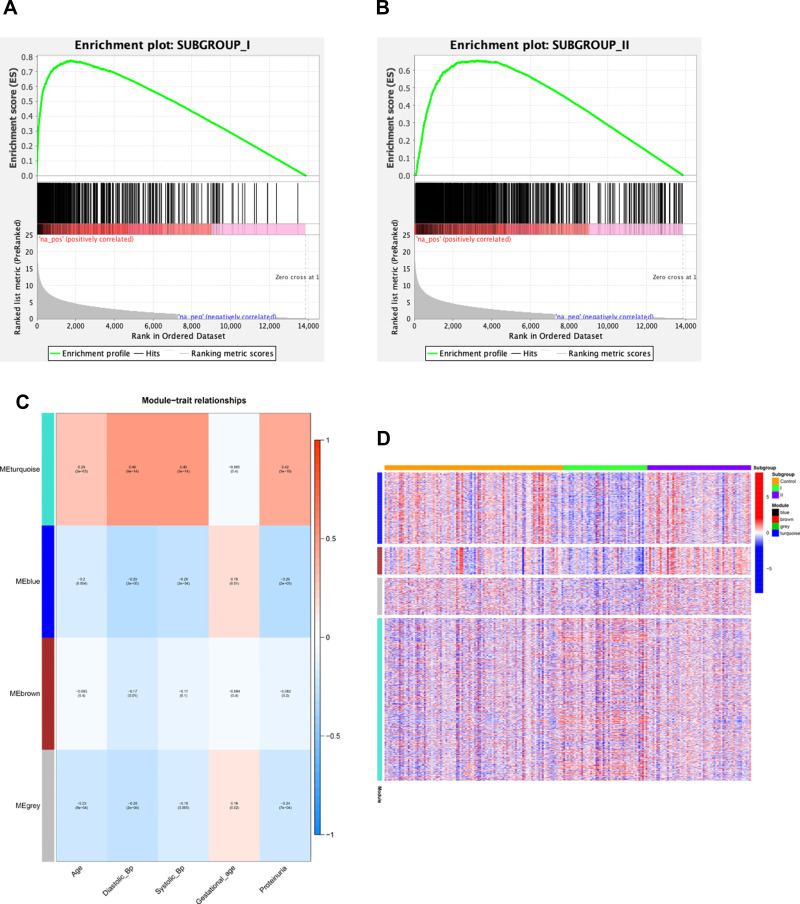
Two PE datasets GSE60438 and GSE75010 were obtained and combined, thereby providing the data of 205 samples in total (100 non-PE and 105 PE samples). After eliminating the batch effect, we grouped and analyzed the integrated data, and further performed GSEA analysis. It was found that the genes in group 1 and group 2 were different from those in normal samples. Moreover, WGCNA analysis revealed that genes in group 1 were up-regulated in turquoise module, including , and , while genes in group 2 were up-regulated in the blue and brown modules. We further conducted GO and KEGG pathway enrichment analyses and found that the differential genes in turquoise module were mainly involved in biological processes such as small molecular catabolic process, while being highly enriched in pathways, including MAPK signaling pathway and Rap1 signaling pathway.
FLT-1 was conventionally used to predict PE risk, and sFLT-1 could also be used as an indicator to evaluate PE treatment effect. As a candidate biomarker for predicting PE, SASH1 may participate in proliferation, migration, invasion and epithelial mesenchymal transformation of human trophoblast cells by regulating MAPK pathway and Rap1 signaling pathway, thus affecting the progression of PE. The mechanism allowing PIK3CB to regulate PE development was not clear, while the gene could be another candidate biomarker for PE risk prediction. This is an exploratory study and our findings were still required verification in further studies.
子痫前期(PE)是一种妊娠特有的多系统疾病,也是孕产妇和围产儿死亡的重要原因。本研究旨在分析已发表数据库中PE患者的胎盘转录数据和临床信息,并预测预防PE的靶基因。
从GEO数据库下载PE患者的临床信息和相应的RNA数据。进行聚类分析以检查不同基因分型基因与临床表现之间的相关性。然后,采用包括GO、KEGG、WGCNA和GSEA在内的生物信息学方法对参与PE发病机制的候选靶基因进行功能表征。
获得并合并了两个PE数据集GSE60438和GSE75010,总共提供了205个样本的数据(100个非PE样本和105个PE样本)。消除批次效应后,我们对整合数据进行分组和分析,并进一步进行GSEA分析。发现第1组和第2组中的基因与正常样本中的基因不同。此外,WGCNA分析显示,第1组中的基因在绿松石模块中上调,包括 、 和 ,而第2组中的基因在蓝色和棕色模块中上调。我们进一步进行了GO和KEGG通路富集分析,发现绿松石模块中的差异基因主要参与小分子分解代谢过程等生物学过程,同时在包括MAPK信号通路和Rap1信号通路在内的通路中高度富集。
FLT - 1传统上用于预测PE风险,sFLT - 1也可作为评估PE治疗效果的指标。作为预测PE的候选生物标志物,SASH1可能通过调节MAPK通路和Rap1信号通路参与人滋养层细胞的增殖、迁移、侵袭和上皮间质转化,从而影响PE的进展。PIK3CB调节PE发展的机制尚不清楚,而该基因可能是另一个用于PE风险预测的候选生物标志物。这是一项探索性研究,我们的发现仍需在进一步研究中进行验证。