Ge Yunhui, Wych David C, Samways Marley L, Wall Michael E, Essex Jonathan W, Mobley David L
Department of Pharmaceutical Sciences, University of California, Irvine, California 92697, United States.
Computer, Computational, and Statistical Sciences Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, United States.
J Chem Theory Comput. 2022 Mar 8;18(3):1359-1381. doi: 10.1021/acs.jctc.1c00590. Epub 2022 Feb 11.
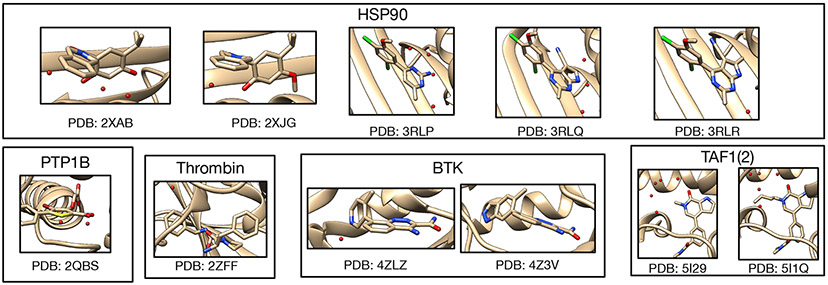
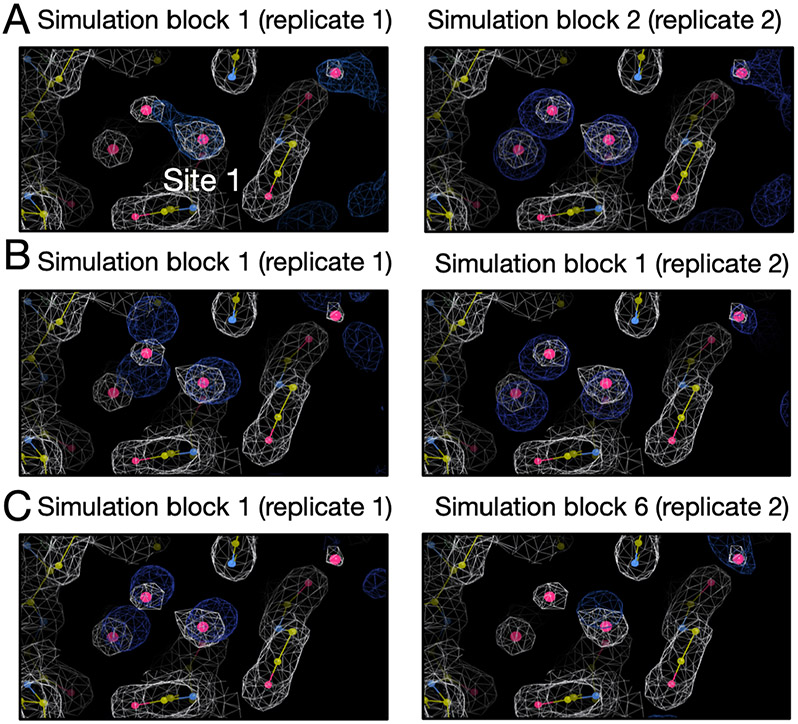
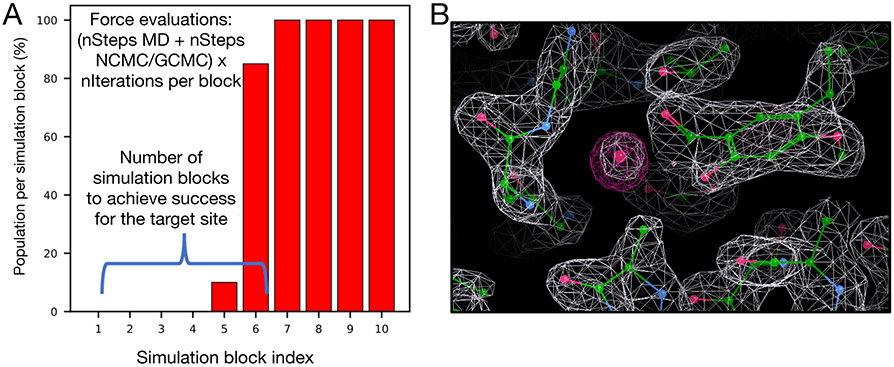
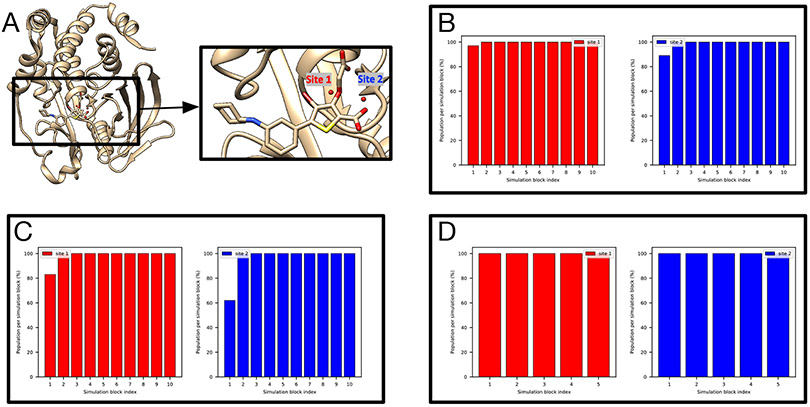
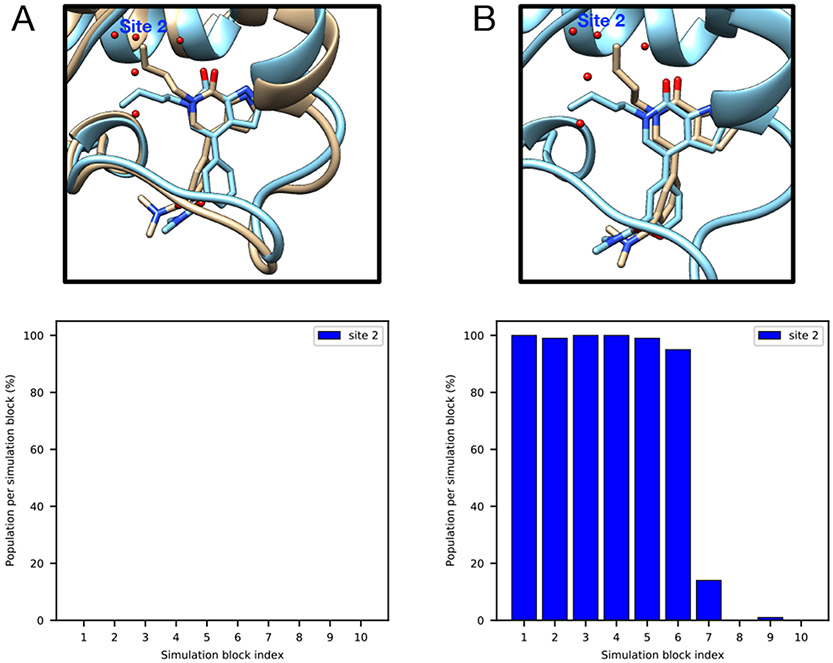
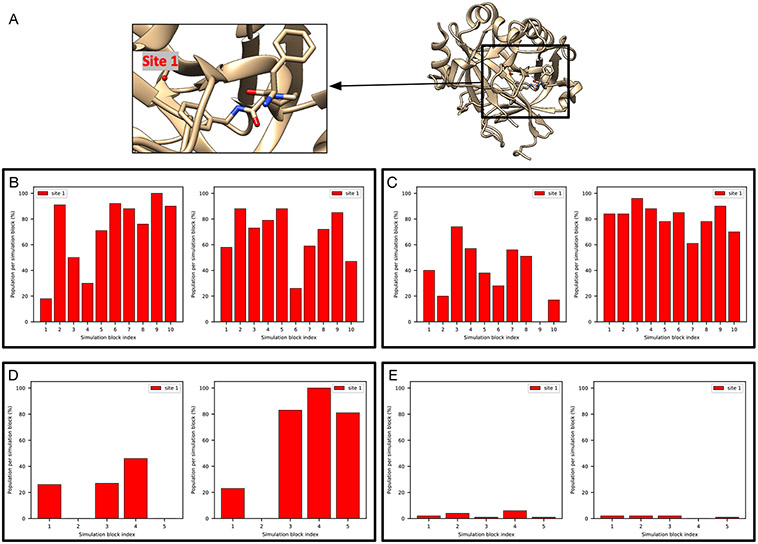
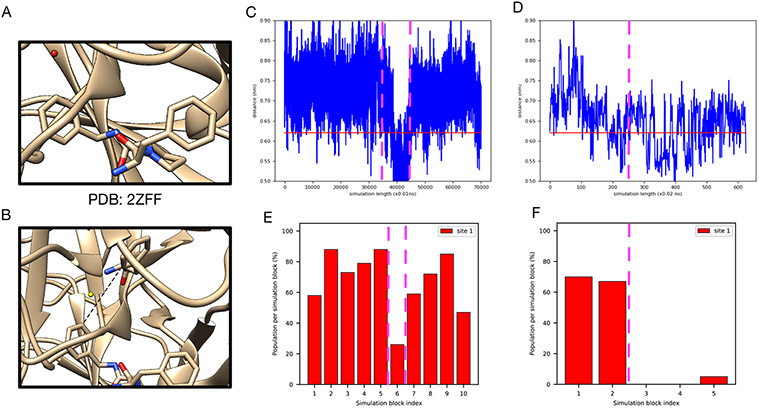
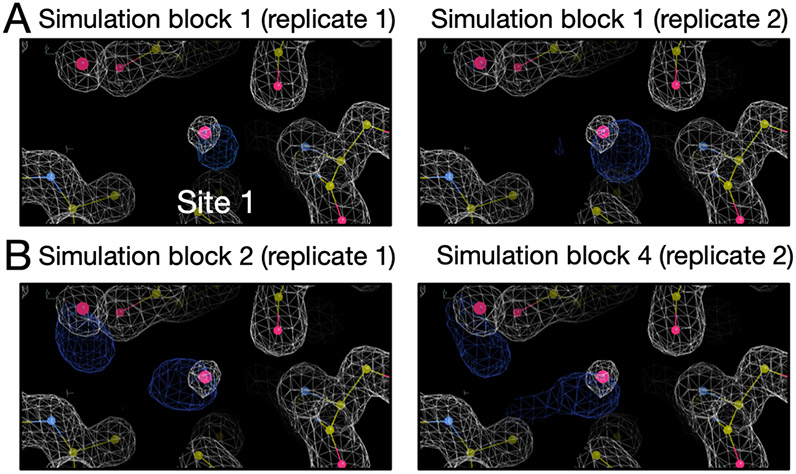
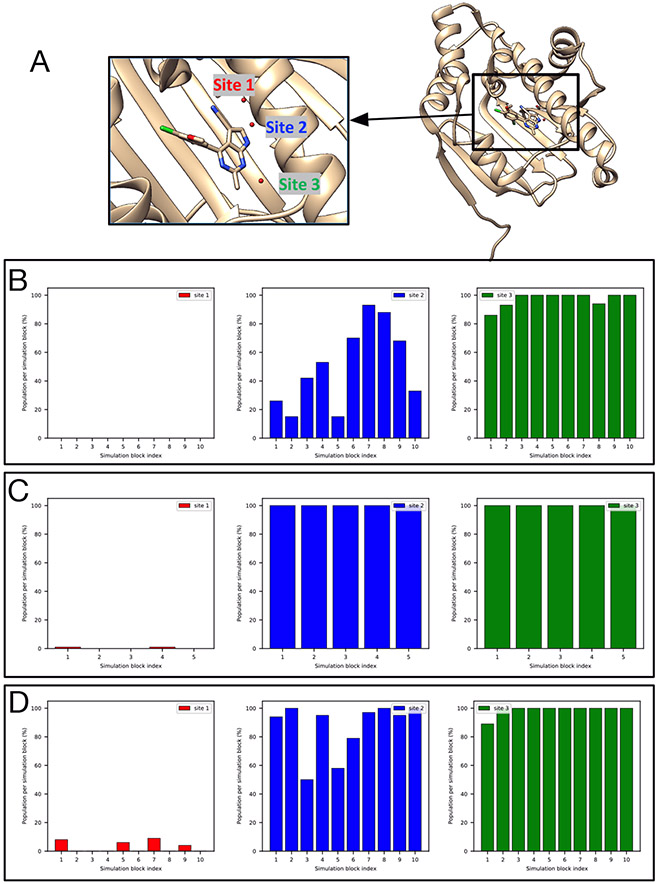
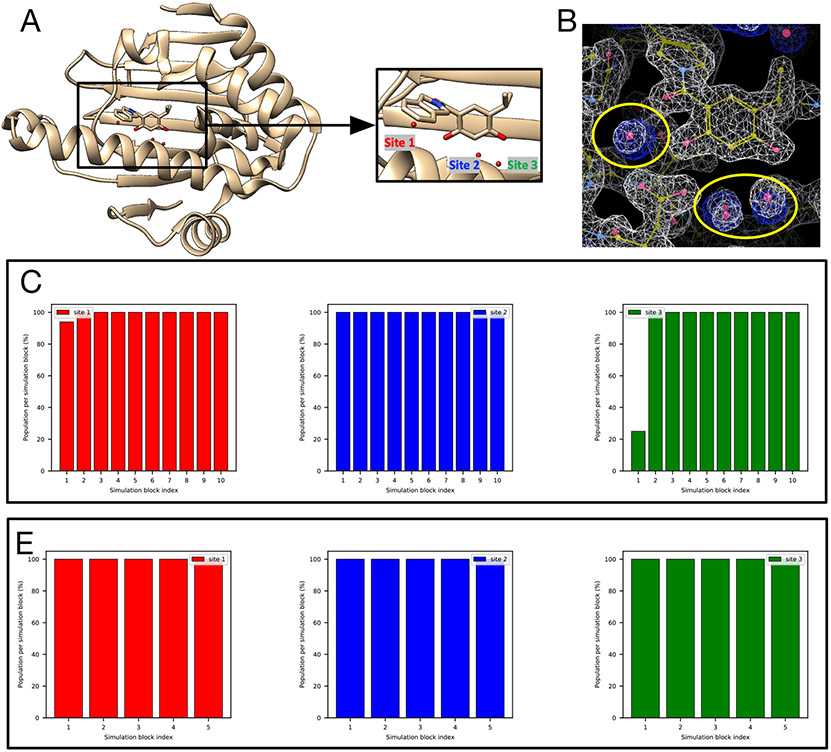
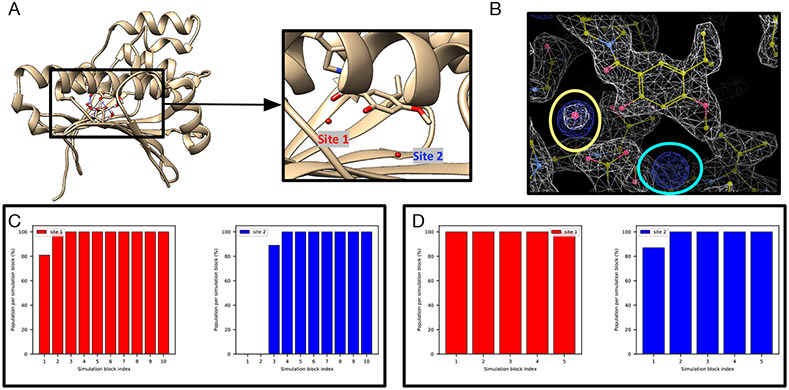
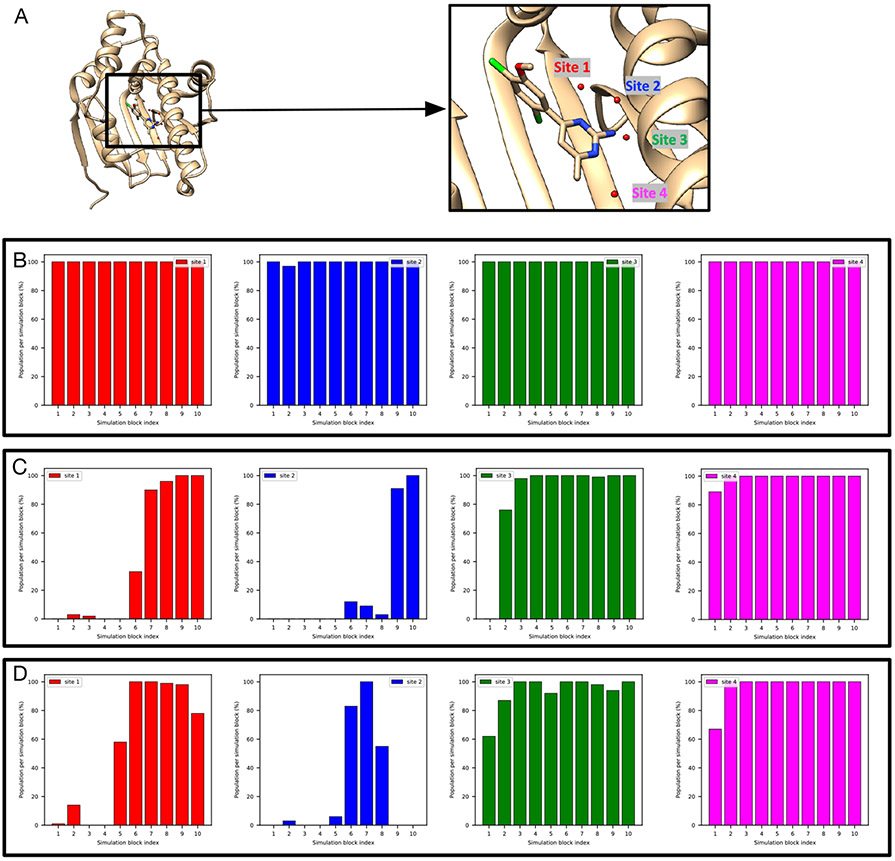
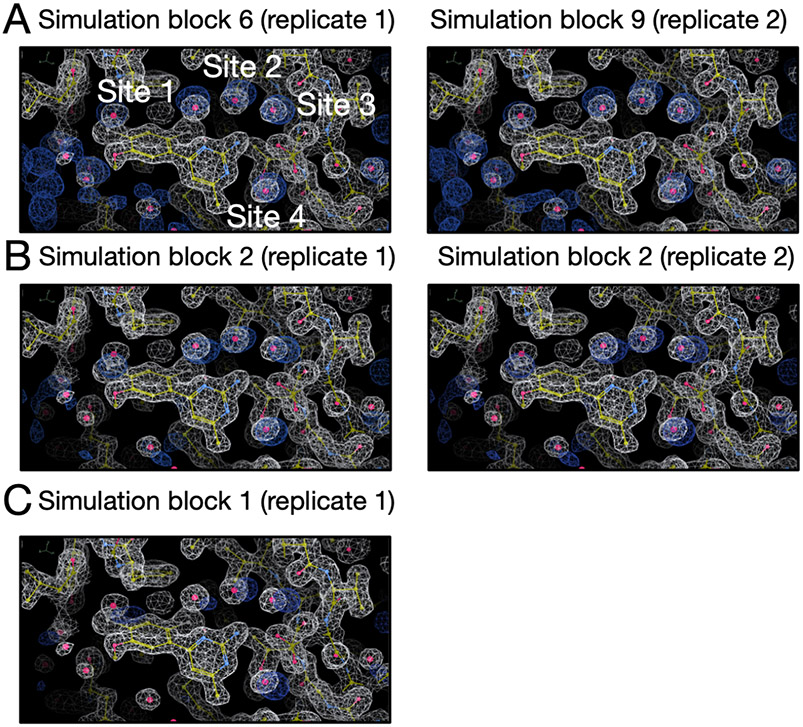
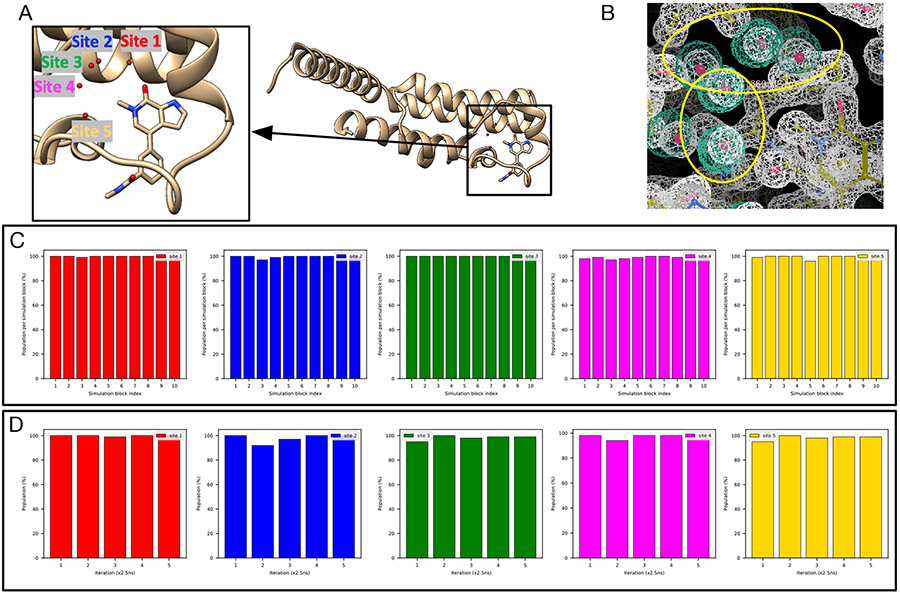
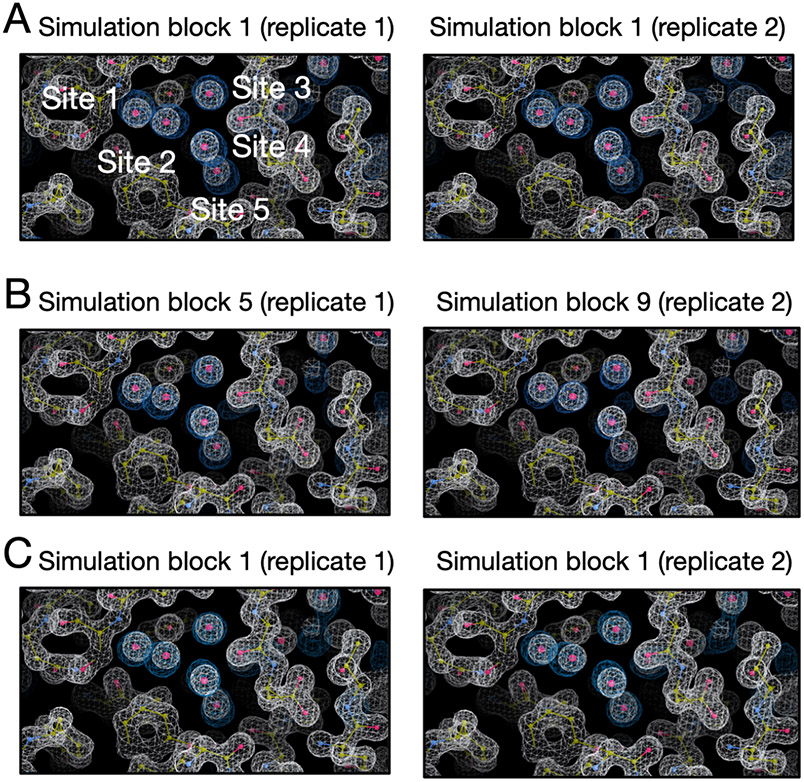
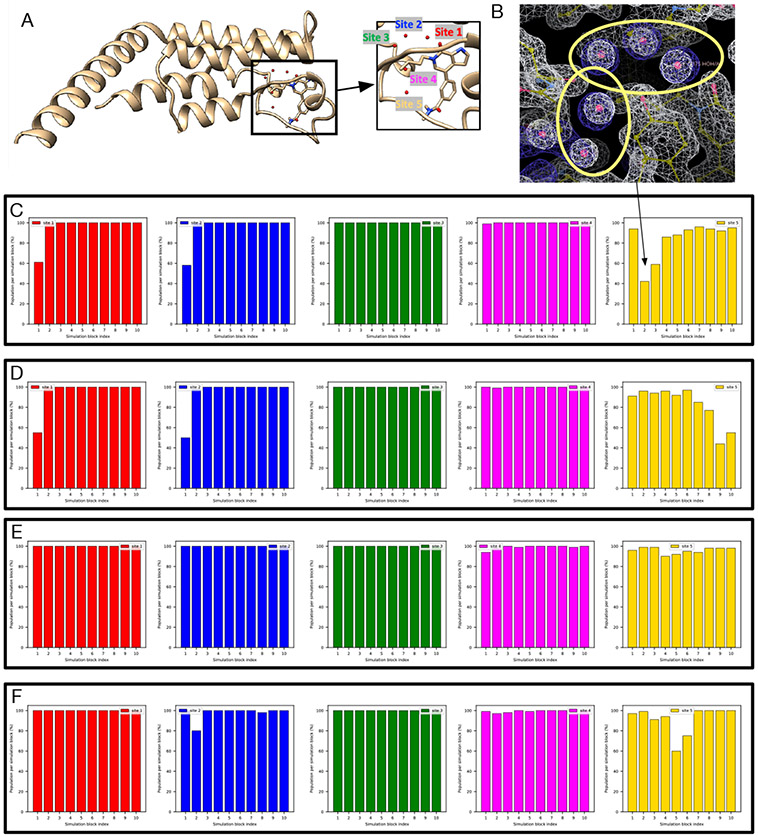
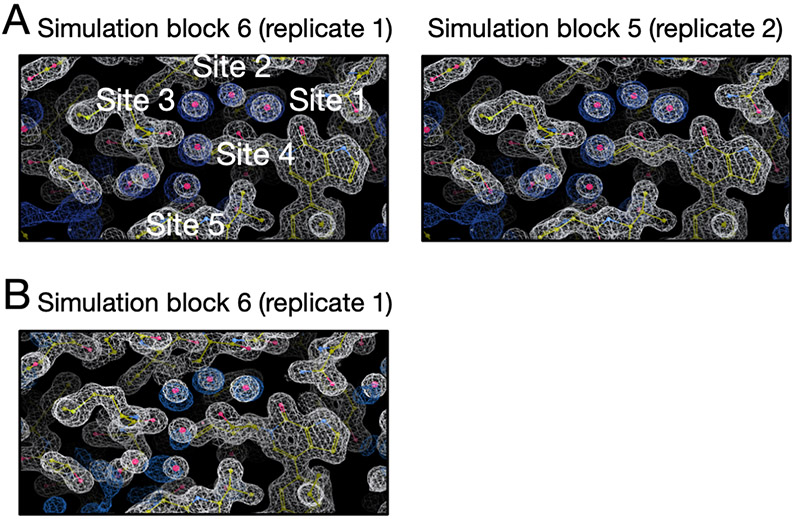
Water often plays a key role in protein structure, molecular recognition, and mediating protein-ligand interactions. Thus, free energy calculations must adequately sample water motions, which often proves challenging in typical MD simulation time scales. Thus, the accuracy of methods relying on MD simulations ends up limited by slow water sampling. Particularly, as a ligand is removed or modified, bulk water may not have time to fill or rearrange in the binding site. In this work, we focus on several molecular dynamics (MD) simulation-based methods attempting to help rehydrate buried water sites: BLUES, using nonequilibrium candidate Monte Carlo (NCMC); , using grand canonical Monte Carlo (GCMC); and normal MD. We assess the accuracy and efficiency of these methods in rehydrating target water sites. We selected a range of systems with varying numbers of waters in the binding site, as well as those where water occupancy is coupled to the identity or binding mode of the ligand. We analyzed the rehydration of buried water sites in binding pockets using both clustering of trajectories and direct analysis of electron density maps. Our results suggest both BLUES and enhance water sampling relative to normal MD and is more robust than BLUES, but also that water sampling remains a major challenge for all of the methods tested. The lessons we learned for these methods and systems are discussed.
水在蛋白质结构、分子识别以及介导蛋白质-配体相互作用中常常发挥关键作用。因此,自由能计算必须充分采样水分子的运动,而这在典型的分子动力学(MD)模拟时间尺度上往往颇具挑战。所以,依赖MD模拟的方法的准确性最终会受到水分子采样缓慢的限制。特别是,当一个配体被移除或修饰时,大量的水可能没有时间在结合位点填充或重新排列。在这项工作中,我们聚焦于几种基于分子动力学(MD)模拟的方法,这些方法试图帮助重新水合埋藏的水位点:使用非平衡候选蒙特卡罗(NCMC)的BLUES;使用巨正则蒙特卡罗(GCMC)的[此处原文缺失相关内容];以及常规MD。我们评估了这些方法在重新水合目标水位点方面的准确性和效率。我们选择了一系列结合位点中水分子数量不同的系统,以及那些水占据情况与配体的身份或结合模式相关联的系统。我们使用轨迹聚类和电子密度图的直接分析来研究结合口袋中埋藏水位点的重新水合情况。我们的结果表明,相对于常规MD,BLUES和[此处原文缺失相关内容]都增强了水分子采样,并且[此处原文缺失相关内容]比BLUES更稳健,但同时也表明水分子采样对于所有测试方法而言仍然是一个重大挑战。我们讨论了从这些方法和系统中吸取的经验教训。