Department of Neuroscience, Mayo Clinic, Jacksonville, FL, USA.
Department of Ophthalmology, The Second Hospital of Jilin University, Changchun, China.
Acta Neuropathol Commun. 2022 Feb 14;10(1):22. doi: 10.1186/s40478-022-01322-x.
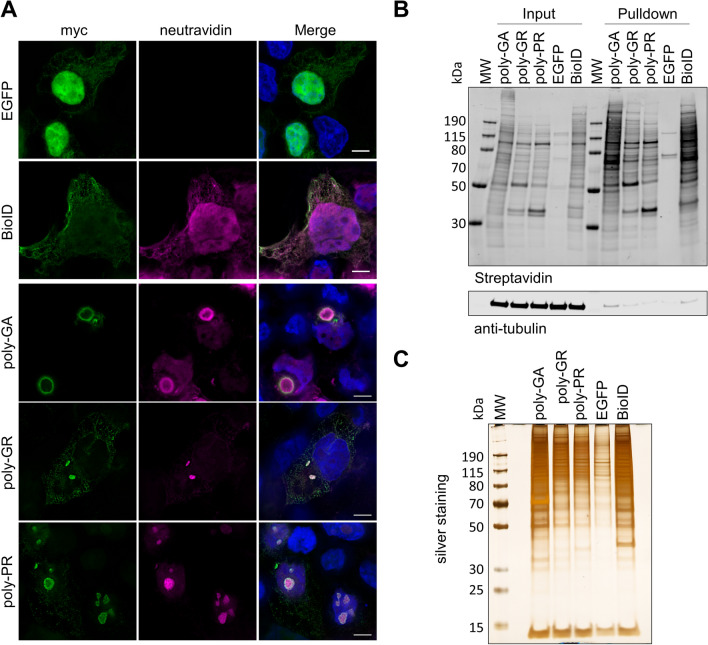
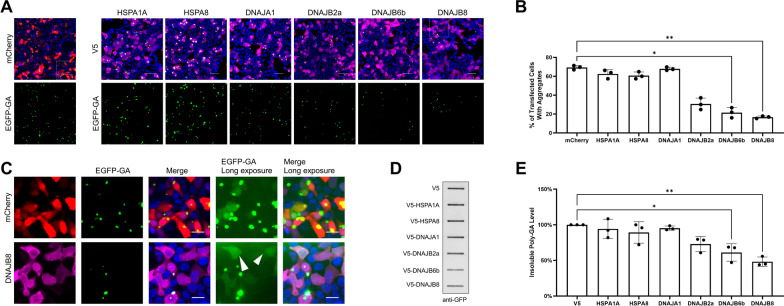
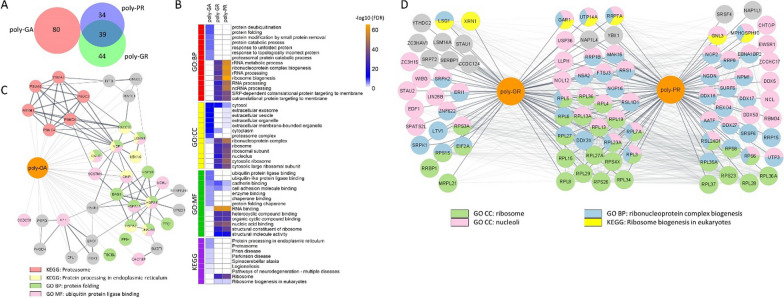
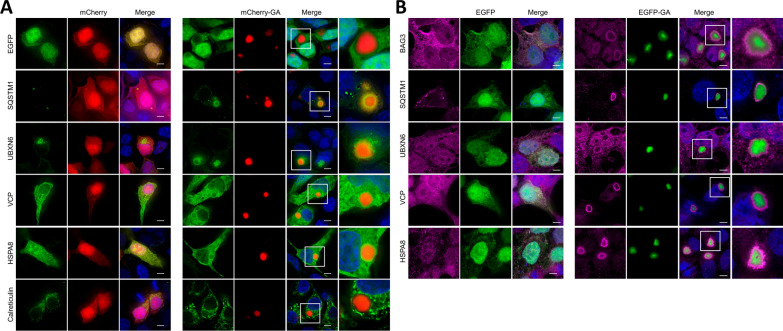
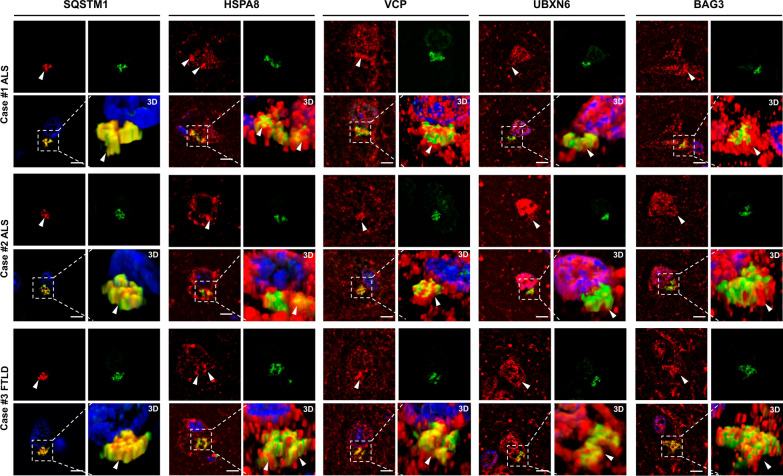
The most common inherited cause of two genetically and clinico-pathologically overlapping neurodegenerative diseases, amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), is the presence of expanded GGGGCC intronic hexanucleotide repeats in the C9orf72 gene. Aside from haploinsufficiency and toxic RNA foci, another non-exclusive disease mechanism is the non-canonical translation of the repeat RNA into five different dipeptide repeat proteins (DPRs), which form neuronal inclusions in affected patient brains. While evidence from cellular and animal models supports a toxic gain-of-function of pathologic poly-GA, poly-GR, and poly-PR aggregates in promoting deposition of TDP-43 pathology and neurodegeneration in affected brain areas, the relative contribution of DPRs to the disease process in c9FTD/ALS patients remains unclear. Here we have used the proximity-dependent biotin identification (BioID) proximity proteomics approach to investigate the formation and collective composition of DPR aggregates using cellular models. While interactomes of arginine rich poly-GR and poly-PR aggregates overlapped and were enriched for nucleolar and ribosomal proteins, poly-GA aggregates demonstrated a distinct association with proteasomal components, molecular chaperones (HSPA1A/HSP70, HSPA8/HSC70, VCP/p97), co-chaperones (BAG3, DNAJA1A) and other factors that regulate protein folding and degradation (SQSTM1/p62, CALR, CHIP/STUB1). Experiments in cellular models of poly-GA pathology show that molecular chaperones and co-chaperones are sequestered to the periphery of dense cytoplasmic aggregates, causing depletion from their typical cellular localization. Their involvement in the pathologic process is confirmed in autopsy brain tissue, where HSPA8, BAG3, VCP, and its adapter protein UBXN6 show a close association with poly-GA aggregates in the frontal cortex, temporal cortex, and hippocampus of c9FTLD and c9ALS cases. The association of heat shock proteins and co-chaperones with poly-GA led us to investigate their potential role in reducing its aggregation. We identified HSP40 co-chaperones of the DNAJB family as potent modifiers that increased the solubility of poly-GA, highlighting a possible novel therapeutic avenue and a central role of molecular chaperones in the pathogenesis of human C9orf72-linked diseases.
最常见的两种遗传和临床病理重叠的神经退行性疾病,肌萎缩侧索硬化症(ALS)和额颞叶痴呆(FTD)的遗传性病因是 C9orf72 基因中存在扩展的 GGGGCC 内含子六核苷酸重复。除了单倍不足和毒性 RNA 焦点外,另一种非排他性疾病机制是重复 RNA 的非规范翻译为五种不同的二肽重复蛋白(DPRs),这些蛋白在受影响的患者大脑中形成神经元包含物。虽然来自细胞和动物模型的证据支持病理性聚 GA、聚 GR 和聚 PR 聚集体的毒性获得功能在促进受影响脑区 TDP-43 病理学和神经退行性变的沉积中具有毒性作用,但 DPRs 对 c9FTD/ALS 患者疾病过程的相对贡献仍不清楚。在这里,我们使用邻近依赖性生物素鉴定(BioID)邻近蛋白质组学方法来研究细胞模型中 DPR 聚集体的形成和集体组成。虽然富含精氨酸的聚 GR 和聚 PR 聚集体的互作组重叠,并富含核仁蛋白和核糖体蛋白,但聚 GA 聚集体与蛋白酶体成分、分子伴侣(HSPA1A/HSP70、HSPA8/HSC70、VCP/p97)、共伴侣(BAG3、DNAJA1A)和其他调节蛋白质折叠和降解的因素(SQSTM1/p62、CALR、CHIP/STUB1)有明显的关联。聚 GA 病理学细胞模型中的实验表明,分子伴侣和共伴侣被隔离到致密细胞质聚集体的外围,导致其从典型的细胞定位中耗尽。在尸检脑组织中证实了它们在病理过程中的参与,其中 HSPA8、BAG3、VCP 及其衔接蛋白 UBXN6 在 c9FTLD 和 c9ALS 病例的额皮质、颞皮质和海马体中与聚 GA 聚集体密切相关。热休克蛋白和共伴侣与聚 GA 的关联促使我们研究它们降低其聚集的潜在作用。我们确定了 DNAJB 家族的 HSP40 共伴侣是有效的调节剂,可增加聚 GA 的溶解度,突出了一种可能的新治疗途径和分子伴侣在人类 C9orf72 相关疾病发病机制中的核心作用。