Wang Qi, Zhang Sheng-Xiao, Chang Min-Jing, Qiao Jun, Wang Cai-Hong, Li Xiao-Feng, Yu Qi, He Pei-Feng
School of Basic Medical Sciences, Shanxi Medical University, Taiyuan, China.
Key Laboratory of Cellular Physiology at Shanxi Medical University, Ministry of Education, Taiyuan, China.
Front Microbiol. 2022 Feb 3;13:799602. doi: 10.3389/fmicb.2022.799602. eCollection 2022.
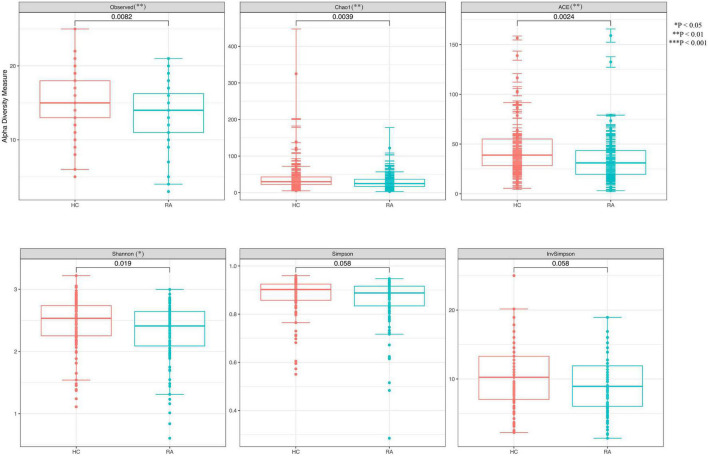
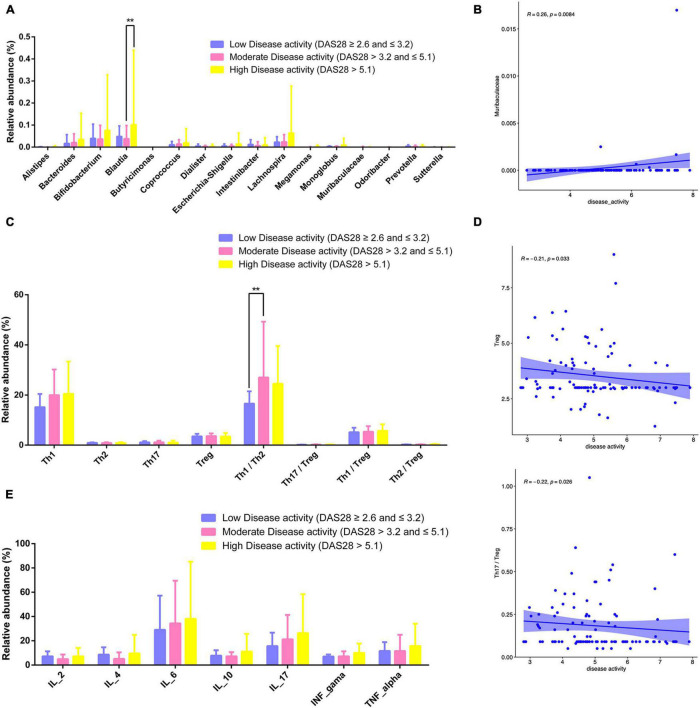
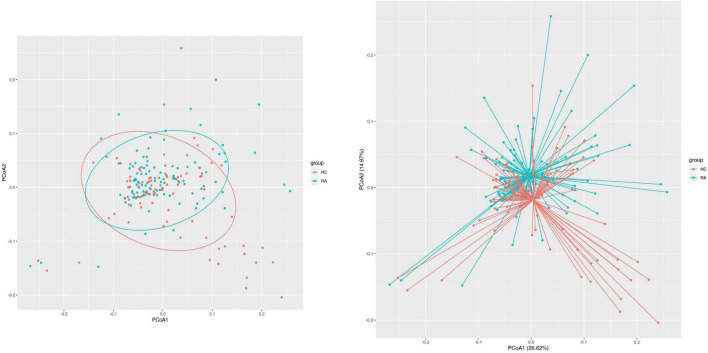
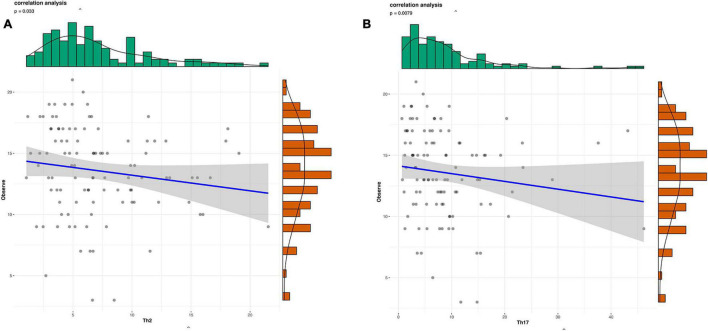
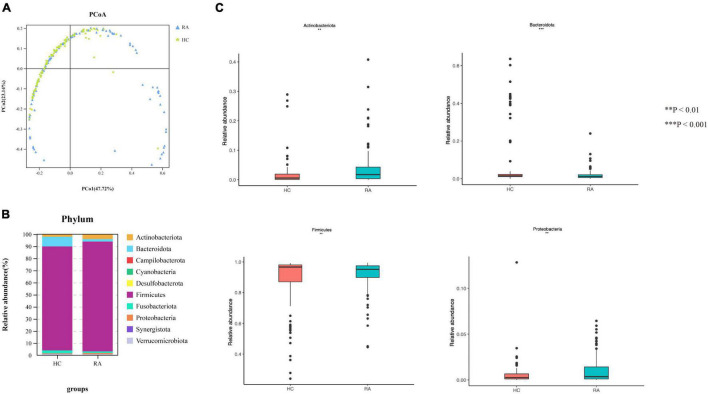
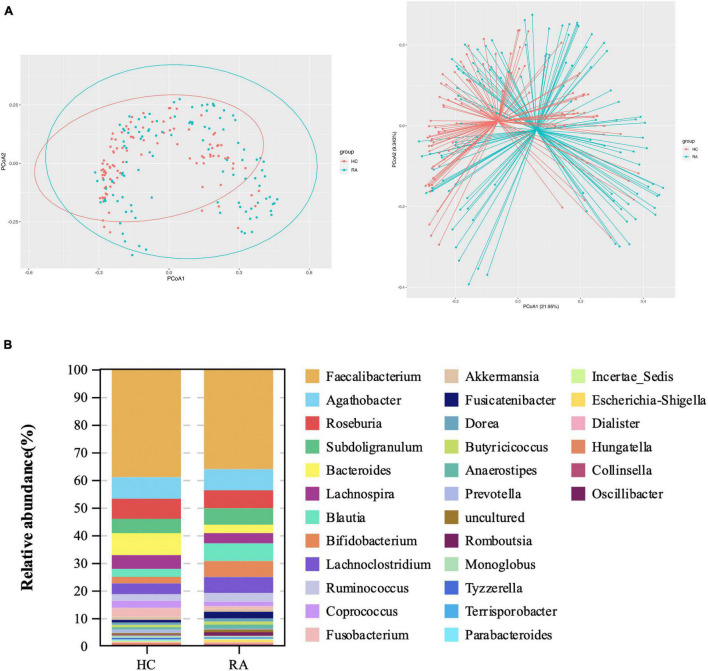
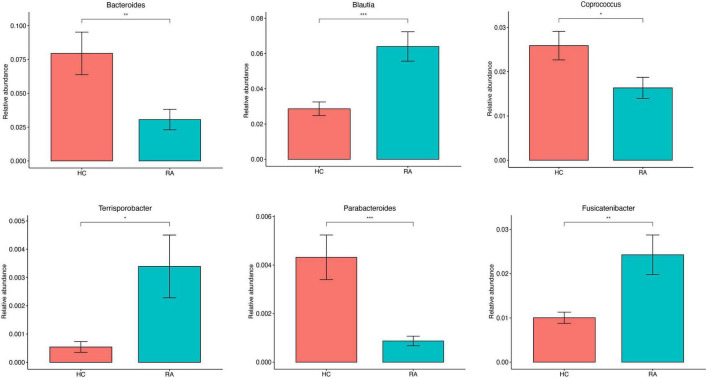
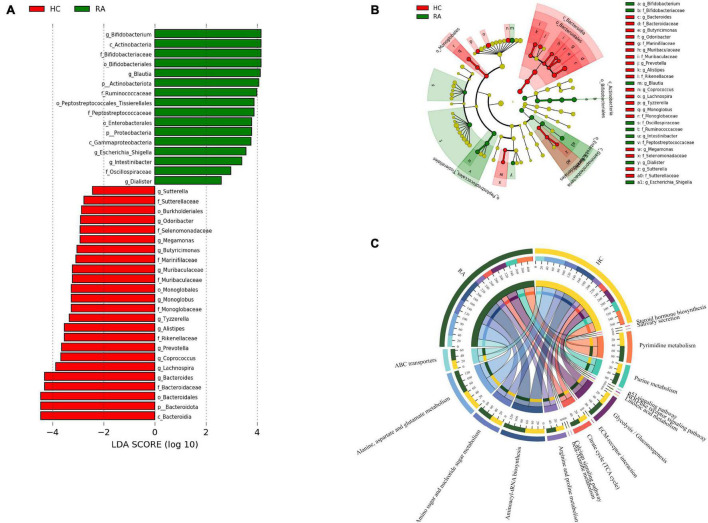
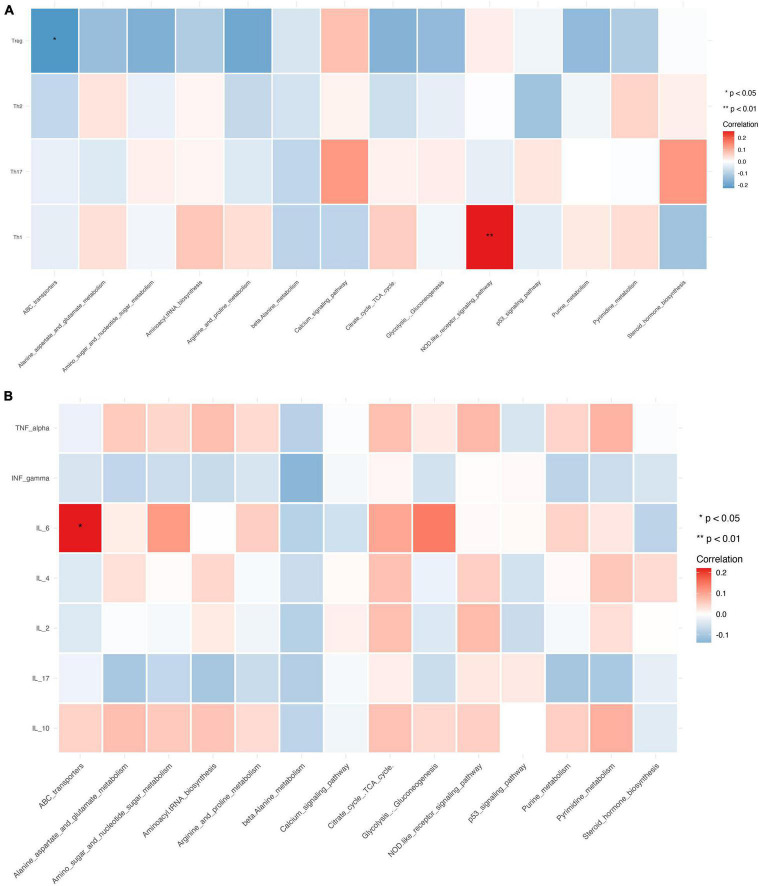
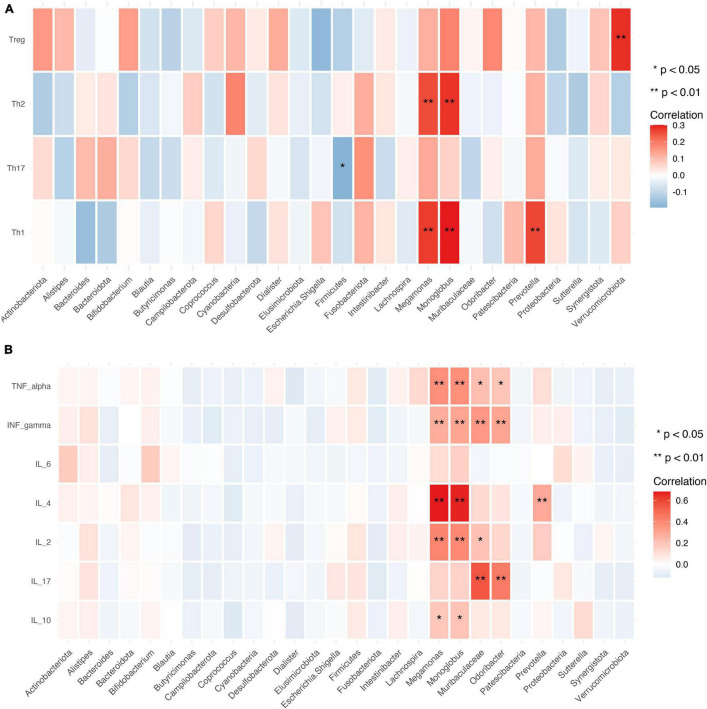
This study investigated the association between intestinal microbiota abundance and diversity and cluster of differentiation (CD)4 T cell subpopulations, cytokine levels, and disease activity in rheumatoid arthritis RA. A total of 108 rheumatoid arthritis (RA) patients and 99 healthy control (HC) subjects were recruited. PICRUSt2 was used for functional metagenomic predictions. Absolute counts of peripheral CD4 T cell subpopulations and cytokine levels were detected by flow cytometry and with a cytokine bead array, respectively. Correlations were analyzed with the Spearman rank correlation test. The results showed that the diversity of intestinal microbiota was decreased in RA patients compared to HCs. At the phylum level, the abundance of Firmicutes, Fusobacteriota, and Bacteroidota was decreased while that of Actinobacteria and Proteobacteria was increased and at the genus level, the abundance of , , and was increased while that of and was decreased in RA patients compared to HC subjects. The linear discriminant analysis effect size indicated that was the most significant genus in RA. The most highly enriched Kyoto Encyclopedia of Genes and Genomes pathway in RA patients was amino acid metabolism. The relative abundance of , , and was positively correlated with CD4 T cell counts and cytokine levels; and the relative numbers of regulatory T cells (Tregs) and T helper (Th17)/Treg ratio were negatively correlated with disease activity in RA. These results suggest that dysbiosis of certain bacterial lineages and alterations in gut microbiota metabolism lead to changes in the host immune profile that contribute to RA pathogenesis.
本研究调查了类风湿关节炎(RA)患者肠道微生物群的丰度和多样性与分化簇(CD)4 T细胞亚群、细胞因子水平及疾病活动之间的关联。共招募了108例类风湿关节炎(RA)患者和99名健康对照(HC)受试者。使用PICRUSt2进行功能宏基因组预测。分别通过流式细胞术和细胞因子珠阵列检测外周血CD4 T细胞亚群的绝对计数和细胞因子水平。采用Spearman等级相关检验分析相关性。结果显示,与HCs相比,RA患者肠道微生物群的多样性降低。在门水平上,厚壁菌门、梭杆菌门和拟杆菌门的丰度降低,而放线菌门和变形菌门的丰度增加;在属水平上,与HC受试者相比,RA患者中 、 和 的丰度增加,而 和 的丰度降低。线性判别分析效应大小表明, 在RA中是最显著的属。RA患者中最高度富集的京都基因与基因组百科全书途径是氨基酸代谢。 、 和 的相对丰度与CD4 T细胞计数和细胞因子水平呈正相关;调节性T细胞(Tregs)的相对数量和辅助性T细胞(Th17)/Treg比值与RA的疾病活动呈负相关。这些结果表明,某些细菌谱系的生态失调和肠道微生物群代谢的改变导致宿主免疫谱的变化,这有助于RA的发病机制。