Khan Kanwal, Jalal Khurshid, Khan Ajmal, Al-Harrasi Ahmed, Uddin Reaz
Dr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan.
HEJ Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan.
Front Microbiol. 2022 Feb 10;12:796363. doi: 10.3389/fmicb.2021.796363. eCollection 2021.
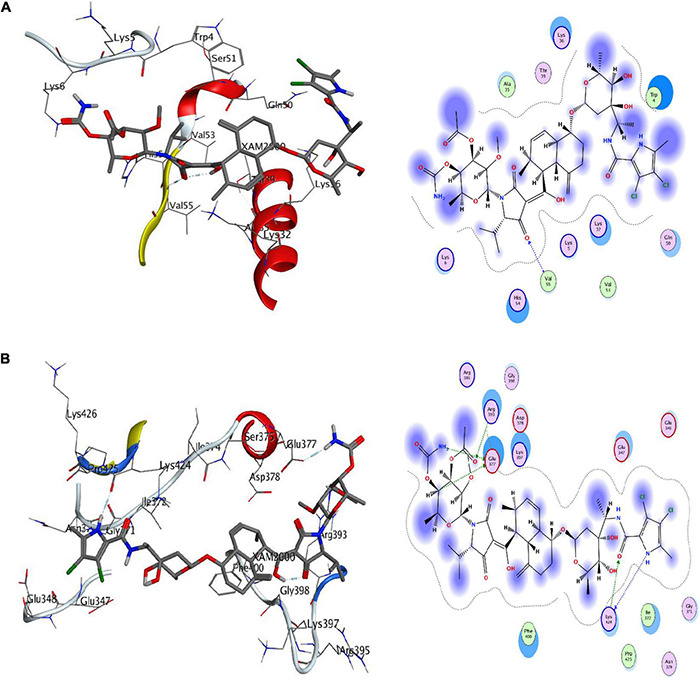
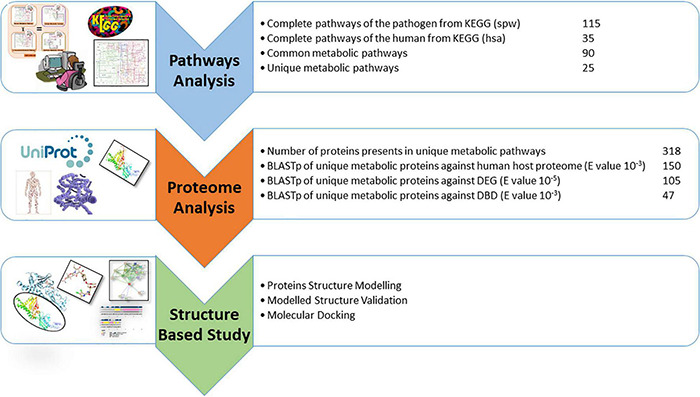
is a notorious pathogen that affects ∼450 million people worldwide and causes up to four million deaths per annum. Despite availability of antibiotics (i.e., penicillin, doxycycline, or clarithromycin) and conjugate vaccines (e.g., PCVs), it is still challenging to treat because of its drug resistance ability. The rise of antibiotic resistance in is a major source of concern across the world. Computational subtractive genomics is one of the most applied techniques in which the whole proteome of the bacterial pathogen is gradually reduced to a limited number of potential therapeutic targets. Whole-genome sequencing has greatly reduced the time required and provides more opportunities for drug target identification. The goal of this work is to evaluate and analyze metabolic pathways in serotype 14 of to identify potential drug targets. In the present study, 47 potent drug targets were identified against by employing the computational subtractive genomics approach. Among these, two proteins are prioritized (i.e., 4-oxalocrotonate tautomerase and Sensor histidine kinase uniquely present in ) as novel drug targets and selected for further structure-based studies. The identified proteins may provide a platform for the discovery of a lead drug candidate that may be capable of inhibiting these proteins and, therefore, could be helpful in minimizing the associated risk related to the drug-resistant . Finally, these enzymatic proteins could be of prime interest against to design rational targeted therapy.
是一种臭名昭著的病原体,全球约有4.5亿人受其影响,每年导致多达400万人死亡。尽管有抗生素(如青霉素、强力霉素或克拉霉素)和结合疫苗(如肺炎球菌结合疫苗),但由于其耐药能力,治疗仍然具有挑战性。该病原体抗生素耐药性的上升是全球关注的一个主要问题。计算减法基因组学是应用最广泛的技术之一,通过该技术,细菌病原体的整个蛋白质组逐渐减少到有限数量的潜在治疗靶点。全基因组测序大大减少了所需时间,并为药物靶点识别提供了更多机会。这项工作的目标是评估和分析该病原体14血清型的代谢途径,以识别潜在的药物靶点。在本研究中,采用计算减法基因组学方法针对该病原体鉴定出47个有效的药物靶点。其中,两种蛋白质(即4-草酰巴豆酸互变异构酶和该病原体中独特存在的传感器组氨酸激酶)被优先作为新型药物靶点,并被选用于进一步的基于结构的研究。所鉴定的蛋白质可能为发现先导药物候选物提供一个平台,该候选物可能能够抑制这些蛋白质,因此有助于将与耐药病原体相关的风险降至最低。最后,这些酶蛋白可能是针对该病原体设计合理靶向治疗的主要关注点。